.............................
THE HONOURABLE MR JUSTICE BUTCHER
Table of Contents:
Introduction. 4
The Factual Framework of the Case. 5
Heart Failure. 5
Ularitide. 6
Cardiorentis. 7
The involvement of IQVIA.. 7
ICH-GCP. 7
The General Services Agreement 8
Change Orders. 12
The Protocol 12
Communications with the FDA in 2012. 18
The Addition of the Co-Primary Efficacy Endpoint 18
The amendment to the Protocol 19
Quality Assurance and the Clinical Quality Agreement (CQA) 21
Other persons and entities involved in the TRUE-AHF trial 23
Investigators. 23
The Executive Committee. 24
The Steering Committee. 24
Documents detailing practices and procedures for the TRUE-AHF trial 24
Monitoring Guideline. 24
The Quality Management Plan. 26
The Protocol Deviation Management Plan. 27
The Medical Monitoring Plan. 30
The Statistical Analysis Plan. 30
The Progress of the Trial and the sites involved. 31
Key Persons involved. 32
Databases and reports. 34
Co-Monitoring visits, audits and inspections. 38
Cardiorentis Remote Monitoring. 42
DSMB Recommendations. 42
The Blind Data Review.. 42
ICH-GCP provisions relevant to Blind Data Review.. 42
The Protocol and SAP as to the Blind Data Review.. 46
The conduct of the Blind Data Review.. 46
The finalised Blind Data Review Plan. 50
The Results revealed. 53
The Response of Cardiorentis and the involvement of SCI 54
The SCI Report 55
The NEJM Article. 58
The NEJM Editorial 65
Other Studies. 66
Nesiritide / ASCEND.. 66
Serelaxin / RELAX. 67
GALACTIC.. 68
The Proceedings Brought 69
North Carolina. 69
IQVIA’s English proceedings. 69
Cardiorentis’s English proceedings. 69
The Issues on the pleadings. 70
Limitation and clarification of Cardiorentis’s case at outset of trial 74
The Factual Witnesses called. 75
The Expert evidence. 76
Agreed facts. 76
The Issues to be decided. 76
The Obligations owed by IQVIA.. 77
General aspects. 77
Medical Monitoring. 78
SDV.. 79
CQA.. 80
Were there breaches of contract by IQVIA which caused there to be eligibility deviations?. 80
Is the number of eligibility deviations itself indicative of breach?. 81
Alleged deficiencies in training. 83
Alleged deficiencies in monitoring. 85
EC3. 87
The nature of the criterion. 87
The facts relevant to EC3 deviations. 87
Cardiorentis’s case. 92
IC6. 95
The facts relevant to IC6 deviations. 96
Cardiorentis’s case. 97
Non-EC3 or IC6 eligibility deviations. 100
SDV.. 102
Alleged breach of CQA.. 103
Other allegations of defective performance. 103
Conclusion as to Breach. 104
The BDR Process. 104
Was the study unreliable or of little use by reason of inclusion of patients with eligibility or other protocol deviations?. 107
Legal principles applicable to claims for wasted costs as damages. 113
The assessment of waste and the relevance of benefits received. 115
Application of legal principles to the facts. 116
The Claim for an Injunction. 117
The Counterclaim.. 118
Overall Conclusion. 121
The Hon. Mr Justice Butcher:
Introduction
1. This case involves a claim by the Claimant (‘Cardiorentis’), a company interested in developing pharmaceutical and biotechnology products, against the Defendants in respect of the conduct of a Clinical Trial, and a counterclaim in respect of unpaid fees relating to that Clinical Trial. The Clinical Trial in question has been given the name TRUE-AHF, standing for Trial to Evaluate the Efficacy and Safety of Ularitide Intravenous Infusion in Patients Suffering from Acute Decompensated Heart Failure.
2. The First Defendant is a major contract research organisation (or ‘CRO’), whose business includes providing clinical trial services, research and other services for the pharmaceutical, medical device and biotechnology industries. It was formerly known as Quintiles Ltd. The Second Defendant is a company incorporated under the laws of North Carolina. It is an indirect parent company of the First Defendant. It was formerly known as Quintiles Inc. In this judgment, save where it is necessary to distinguish them, I will refer to the Defendants as ‘IQVIA’. In many of the documents to which reference is made, the name used is ‘Quintiles’, and I have not sought to change these.
3. This case involved looking in detail at the contractual arrangements for, and the conduct and results of, the TRUE-AHF study. A great deal of the factual material relating to the origin, background to, course of and results of the Trial were not in issue or not significantly in issue. To understand the issues in the case it is necessary to understand many of these matters. Accordingly, the next section of this judgment contains a summary of them. Its contents were largely common ground. To the extent it was not, it represents my findings based on the documentary evidence.
Heart Failure
4. Heart failure (‘HF’) is a syndrome encompassing a variety of cardiac diseases which progress to imposing on the heart injury, cellular loss, overload and change in dimensions making the heart less efficient in supporting blood supply to other organs and thereby making them dysfunctional. The disease usually progresses chronically, with some relatively, and seemingly stable, ‘compensated’ periods, albeit with a continuous slow decline of health and with a permanent risk of sudden death. Occasionally the disease enters ‘decompensated’ episodes of rapid worsening, called ‘Acute Decompensated Heart Failure’ (‘ADHF’), which is a life-threatening condition, occasionally complicated by death, and usually requiring hospital admission for appropriate care.
5. There are multiple causes of decompensation. Some relate to aggravation of the underlying cardiac disease, such as myocardial infarction, angina and hypertension; others to drug interactions. Non-compliance with a patient’s drug therapy or dietary restrictions are also frequent causes. Excessive fatigue related to intense exercise, the advent of cardiac arrhythmia or of pulmonary infection can be causes of excessive work being imposed on the heart and can precipitate decompensated HF.
6. The major symptoms of HF are related to congestion: ie fluid accumulation in the body and mainly in the lungs related to poor cardiac output and reduced renal excretion of water and sodium. The cardinal symptom is shortness of breath, or dyspnoea, related to lung oedema. ADHF is characterised by dyspnoea at rest. Dyspnoea which is aggravated when the patient is in the recumbent position is an indication that the patient is severely affected. ADHF may also be diagnosed by radiological evidence. In cases of ADHF chest X-rays will typically show bilateral fluid accumulation.
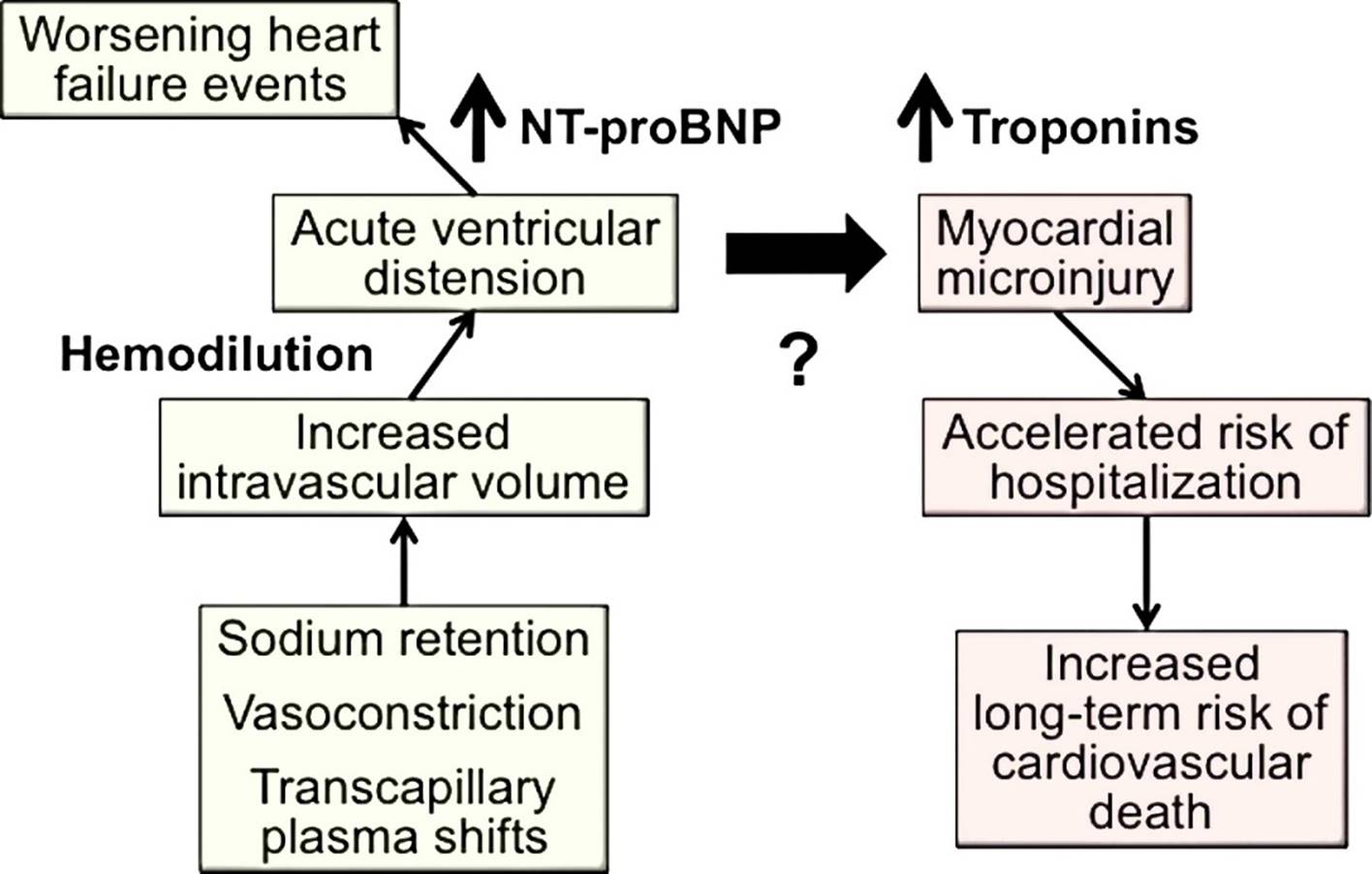
7. Brain natriuretic peptide (‘BNP’) and N-terminal pro BNP (‘NT-pro BNP’) are molecules from the same natriuretic peptide family, which are produced in the heart. The production of these hormones is stimulated when stress is imposed on the heart. In normal conditions, such production helps the heart cope with excessive exercise, helping blood vessels to dilate (vasodilation), unloading the heart and enabling sodium and water to be more readily excreted from the kidneys, thus preventing congestion. In HF, permanent excessive stress on the cardiovascular system produces an excessive chronic increase of these hormones, and this is further amplified during acute episodes such as ADHF. There is a direct relationship between blood levels of the natriuretic peptides, BNP and NT-pro BNP, and the severity of heart failure. They are accordingly referred to as ‘biomarkers’. The presence of BNP and NT-pro BNP in the blood above a certain level is an indicator of the degree of HF; a drop in the level of BNP / NT-pro BNP indicates that the heart is under less stress and there is less congestion.
8. Cardiac troponins are a group of proteins which help regulate the contraction of the cardiac muscle. Where myocardial cells are injured troponin is released into the blood. This makes troponin a useful biomarker of heart injury.
9. The demographic distribution of patients with acute HF follows that of chronic HF. It mostly affects the elderly. Women and men are equally affected. More men than women are, however, affected by reduced ejection HF (ie where the heart has difficulty pumping rather than filling) because the dominant cause of this is coronary artery disease which affects men more than women.
10. Acute HF is a global public health problem and is one of the leading causes of hospitalisation (and mortality) worldwide, especially in the USA.
Ularitide
11. The drug which was the subject of the TRUE-AHF trial at the centre of these proceedings is ularitide. Ularitide is a chemically synthesized analogue of urodilatin, a human endogenous natriuretic peptide that is expressed in the kidney and regulates renal sodium and water excretion. The main pharmacological effects of exogenously administered ularitide are vasodilation of renal, pulmonary and coronary arteries, diuresis and natriuresis, and inhibition of the renin-angiotensin-aldosterone system.
12. Ularitide was first found by Professor Markus Meyer and others. In investigating its properties, they identified that ularitide might be an effective treatment for ADHF. As such, in 2001 Professor Meyer and others formed CardioPep Pharma GmBH (‘CardioPep’), with the purpose of developing ularitide for the treatment of that condition.
13. To obtain regulatory approval for new drugs it is necessary to conduct clinical trials. The regulatory approval process varies slightly by country and region, but the overall process, involving Phase I, Phase II and Phase III trials, is similar. Phase I trials for new products are undertaken on a small number of healthy volunteers and focus on an assessment of the safety of the product by investigating its side effects when administered to healthy human beings. Phase II trials for new products are undertaken on patients affected with the indication for which the new product is being investigated as a potential treatment, and focus on an initial assessment of the effectiveness of the product, whilst continuing to evaluate the treatment’s safety. Phase III trials are used to evaluate both safety and efficacy of a product or treatment in a much larger population that is more representative of the ‘real world’ environment in which a product may be used, or at different doses, depending on the trial design.
14. Phase I trials in respect of ularitide were conducted in the 1990s. These were followed by a Phase IIa trial called SIRIUS I (Safety and efficacy of an Intravenous placebo controlled Randomised Infusion of Ularitide in a prospective double-blind Study), conducted between January 2001 and February 2003. The results from SIRIUS I were published in the American Heart Journal in December 2005, and the main conclusion was that ularitide might be a new agent for the therapy of ADHF.
15. Between February 2003 and October 2004 CardioPep conducted a Phase IIb trial to evaluate the efficacy of ularitide at different doses as a treatment for patients with symptomatic decompensated chronic HF. This trial was known as SIRIUS II. Its results were published in the European Heart Journal in October 2006. The main conclusion was that ‘ularitide lowered cardiac filling pressures and improved dyspnoea without apparent early deleterious effects on renal function … These results suggest that Ularitide may play a role in the management of [decompensated HF].’
Cardiorentis
16. In about 2010, CardioPep agreed to partner with Dr Johannes Holzmeister, a cardiologist and physician, at the time a specialist in the Cardiology Department of University Hospital, Zurich. At about this point Cardiorentis was set up, and CardioPep became a wholly-owned subsidiary of Cardiorentis, and the rights to develop ularitide were licensed from CardioPep to Cardiorentis. Dr Holzmeister began, with others, to seek funding for a Phase III trial for ularitide. At some point thereafter, Dr Holzmeister met Mr Frank Binder, a part of the Merck family, who expressed an interest in investing in the project, and who proceeded to acquire Cardiorentis and to inject funds into the company for the purposes of the Phase III trial.
The involvement of IQVIA
17. Cardiorentis, as a relatively small organisation, could not conduct a Phase III trial on its own, and needed to employ a CRO. On 26 August 2011, an Authorisation to Proceed Agreement (or ‘APA’) was entered into between a company called Euronyme GmbH (‘Euronyme’), of which Dr Holzmeister was CEO, and Quintiles Ltd (now IQVIA Ltd). Under this APA, the parties acknowledged that they were in the course of negotiating a comprehensive agreement regarding the proposed Phase III study of ularitide as a treatment for acute HF. In the interim IQVIA was to provide Euronyme with various services in exchange for specified fees. On 16 November 2011 a Novation and Amendment Agreement was executed by Euronyme, IQVIA and Cardiorentis to transfer Euronyme’s rights under the APA to Cardiorentis.
18. Cardiorentis and IQVIA proceeded to negotiate the terms of the agreement under which IQVIA would provide services in respect of the Phase III trial in prospect.
ICH-GCP
19. The ethical and professional framework within which this agreement was agreed included in particular the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (‘ICH’) Efficacy Guidelines, particularly the Good Clinical Practice (‘GCP’) Guideline E6, which serve as the foundation on which clinical trials are conducted. The ICH Efficacy Guidelines, including in particular Guidelines E6 and E8, together with any other regulatory requirements for a given geographic location, provide a framework for clinical trials to ensure the protection of human subjects and the integrity of the study data.
20. Specifically, ICH GCP E6 (R1) was the relevant version in place at the material time of the TRUE-AHF trial. An updated version (R2) was published in 2016. Some key concepts contained in E6 (R1) were:
(1) Any study must have a ‘Sponsor’, namely the person / entity on whose behalf the drug is being investigated. E6 (R1) para 1.53 defines a Sponsor as ‘an individual, company, institution, or organization which takes responsibility for the initiation, management, and/or financing of a clinical trial’. Cardiorentis was the Sponsor of the TRUE-AHF trial.
(2) The Sponsor is responsible for ‘implementing and maintaining quality assurance and quality control systems with written SOPs to ensure that trials are conducted and data are generated, documented (recorded), and reported in compliance with the protocol, GCP, and the applicable regulatory requirement(s).’ (E6 (R1) para 5.1.1)
(3) A Sponsor may ‘transfer any or all of the Sponsor's trial-related duties and functions to a CRO, but the ultimate responsibility for the quality and integrity of the trial data always resides with the Sponsor. The CRO should implement quality assurance and quality control.’ (E6 (R1) para 5.2.1). Cardiorentis engaged IQVIA as CRO to perform certain trial-related duties and functions.
(4) The duties transferred should be specified in writing. (E6 (R1) para 5.2.2).
(5) Where a function is transferred, references in the Guidelines to the Sponsor ‘also apply to a CRO to the extent that a CRO has assumed the trial related duties and functions of a Sponsor’. (E6 (R1) para 5.2.4)
21. Parameters for statistical analysis and the content and format of clinical study reports are included in ICH E3 and ICH E9. Various of these provisions will be referred to below.
22. On 29 May 2012, IQVIA and Cardiorentis entered into a General Services Agreement to provide for a financial reconciliation with sums paid by Cardiorentis to IQVIA under the APA and to agree, within the framework of the ICH GCP Guidelines, upon further services which IQVIA would provide in respect of the TRUE-AHF study. No detailed consideration of this Agreement is necessary because it was soon superseded by another General Services Agreement dated 30 August 2012.
23. The General Services Agreement of 30 August 2012 (to which I will refer as ‘the GSA’) is the main contract governing the relations between Cardiorentis and IQVIA relevant to this claim. The GSA was subject to English law.
24. The GSA contained a number of terms which are agreed or said by the parties to be relevant to the current issues. Recital B to the GSA provided as follows:
‘B. Quintiles [i.e. IQVIA UK] and Sponsor [i.e. Cardiorentis] are the parties to a General Services Agreement dated 29 May 2012 and made between Sponsor and Quintiles (“First GSA”); this follows an Authorization to Proceed Agreement dated 26 August 2011 and made between Euronyme GmbH (“Euronyme”) and Quintiles and the subsequent novation of Euronyme’s rights and obligations to Sponsor under the Novation and Amendment Agreement made between the parties hereto and Euronyme on 7 November 2011. The said Authorization to Proceed Agreement as novated and amended is referred to below as “the ATP”.’
25. Clause 1.0 of the GSA provided:
‘Services to be Provided. The services to be performed hereunder (the “Services”) shall be specified in the Scope of Work attached hereto as Attachment 1 (including some services already carried out under the ATP and First GSA). Any regulatory obligations or other responsibilities not specifically transferred in this Agreement shall remain the responsibility of Sponsor.’
26. The Scope of Work in Attachment 1 included the following:
(1) Investigator Meeting and CRA [Clinical Research Associate] Training: Investigator Meeting Planning and Coordination:
‘Quintiles will be responsible for planning and coordinating 4 2-day investigator meeting(s) for up to 194 sites with up to 452 attendees, including sites (388), Sponsor (15), and Quintiles (49) attendees participating. In collaboration with meeting planners to be contracted ad hoc for the events.’
(2) Clinical Operations Detail: Site Initiation Visits
‘Quintiles will perform 194 site initiation visits based on 194 total sites’.
(3) Clinical Operations Detail: Interim Monitoring Visits
‘Quintiles will conduct appropriate number of interim monitoring visits based on 204,926 total CRF [Case Report Form] pages with an average review time of 4 minutes per CRF page. 100% SDV [viz Source Data Verification, explained below] is assumed for budget purposes, however an SDV planned (sic) for reduced SDV will be issued during the study, in order to use the additional time to SDV Endpoint and SAE data’
(4) Medical Monitoring Detail: Medical Monitoring
‘Quintiles’ Medical Advisor will provide medical monitoring as follows. Sponsor will be responsible for all other medical monitoring activities’
(5) Medical Monitoring Detail: Medical Input for Protocol Development
‘Quintiles’ Medical Advisor will provide medical consultation to Sponsor’s and Quintiles’ medical writer during protocol development.’
(6) Medical Monitoring Detail: CRA Training
‘Quintiles’ Medical Advisor will provide 3 8 hour CRA training sessions’
(7) Medical Monitoring Detail: CRA and Site Support
‘Quintiles’ Medical Advisor will provide medical monitoring activities throughout the start-up, recruitment, treatment, and close-out phases of the study during normal business hours
Quintiles Medical Advisor will not diagnose or propose treatment for patients. All decisions associated with patient care will be made by the investigators’
(8) Biostatistics Detail: Blinded Data Review Meeting
‘Quintiles’ biostatistician will attend 1 8-hour blinded data review meeting via teleconference with Sponsor before the database is locked and the project is unblinded to resolve all outstanding data and analytical issues. Specifications of review materials (e.g. blinded data TLFs) are described above as intermediate statistical deliverables’
27. Clause 2 of the GSA made provision for the payment of fees and expenses. It provided, in part:
‘Payment of Fees and Expenses. All monies paid under the ATP and the First GSA will be applied towards and reconciled with the payments specified in this Agreement. Sponsor will pay Quintiles the fees, expenses and pass-through costs in accordance with the budget (Attachment 2) and payment schedule (Attachment 4) attached...
With the exception of any invoices for prepayment or advances and investigator invoices, which are due within ten (10) calendar days upon receipt, all other invoice payments shall be made to Quintiles within thirty (30) days of receipt... Quintiles reserves the right to impose, and Sponsor agrees to pay if imposed by Quintiles, interest in an amount equal to two percent (2%) above the one month base interest rate established by the Bank of England per month … of all undisputed amounts owing hereunder, which are outstanding thirty (30) or more days from the due date of the invoice…
Quintiles is also providing Sponsor an additional pricing incentive for this General Services Agreement related to achievement of “Key Milestones” or “Performance Targets” as described herein (the “Service Delivery Excellence Program”). Quintiles will apply a Service Delivery Excellence Program reduction set forth herein as fifteen percent (15%) off Quintiles’ professional labor fees for any project management, regulatory, clinical monitoring, medical services, medical writing, data management, biostatistics or pharmacovigilance services performed by Quintiles under this General Services Agreement... Quintiles will have an opportunity to earn back some or all of the Service Delivery Excellence Program reduction by meeting or exceeding one or more of the five (5) Key Milestones or Performance Targets set forth below... ‘
28. The Key Milestones included, as 4, ‘Statistical Analysis (Top Line Result)’, the Target Achievement being 13 days after Database Lock, and the Percentage of Fees for Quintiles’ Traditional Services Available for Earn Back being 3%. The GSA further provided:
‘If Quintiles shall fail to meet one or more of the targets then Quintiles will not earn back the applicable proportion of the discount …’
29. Clause 5 of the GSA provided for Change Orders. It was, in part, in these terms:
‘Change Orders. Any change in the details of this Agreement or the assumptions upon which this Agreement is based … may require changes in the budget and/or time lines, and shall require a written amendment to the Agreement (a “Change Order”)…’
30. Clauses 7 and 8 of the GSA provided in part:
‘7.0 a) Ownership and Inventions. Excluding Quintiles’ Property (defined below), all data and information generated or derived by Quintiles as the result of Services performed by Quintiles under this Agreement and which are provided by Quintiles to Sponsor as deliverables under this Agreement shall be and remain the exclusive property of Sponsor…
8.0 Records and Materials At the completion of the Services by Quintiles, all materials, information and all other data owned by Sponsor, regardless of the method of storage or retrieval, shall be delivered to Sponsor in such form as is then currently in the possession of Quintiles, subject to the payment obligations set forth in Section 2.0 herein. Alternatively, at Sponsor’s written request, such materials and data may be retained by Quintiles for Sponsor for an agreed-upon time period, or disposed of pursuant to the written directions of Sponsor…. Nothing in this Agreement shall be construed to transfer from Sponsor to Quintiles any FDA [ie the US Food and Drug Administration] or regulatory record-keeping requirements unless such transfer is specifically provided for in the applicable Transfer of Obligations Form.’
31. Clause 10(a) of the GSA stipulated that IQVIA’s services would be performed ‘with the standard of care customary in the contract research organization industry. Quintiles’ standard operating procedures will be used in performance of the Services, unless otherwise specifically stated in the Scope of Work.’
32. Clause 11 provided, in part
‘Relationship with Investigators
…
The parties acknowledge and agree that, except as to employees of Quintiles or its affiliates, Investigators shall not be considered the employees, agents, or subcontractors of Quintiles. All Investigators shall exercise their own independent medical judgment. Quintiles’ responsibilities with respect to Investigators shall be limited to those responsibilities specifically set forth in this Agreement….’
33. Clause 16 provided for limitations of liability, as follows:
‘16.0 Limitation of Liability.
(a) Neither Quintiles, nor its affiliates, nor any of their respective directors, officers, employees, subcontractors or agents shall have any liability (including without limitation, contract, negligence and tort liability) for any loss of profits, opportunities or goodwill or any type of indirect or consequential damages in connection with this Agreement or the Services performed by Quintiles.
(b) In no event shall the collective, aggregate liability (including without limitation, contract, negligence and tort liability) of Quintiles or its affiliates, directors, officers, employees, subcontractors or agents under this Agreement exceed the amount of fees actually received by Quintiles from Sponsor under this Agreement.
…’
34. As provided for by Clause 5.0 of the GSA, a series of Change Orders were agreed during the course of the contract. Although it involves looking ahead chronologically, it is convenient to refer here to what those Change Orders were. There were seven of them in all of which the most relevant were Change Orders 1, 2, and 5-7.
35. Change Order 1 was dated 22 May 2013, but signed by Cardiorentis on 23 May 2013, and by IQVIA on 4 June 2013. It provided for an increase of about Eur 4.2 million in IQVIA’s budgeted pass-through costs (to be paid by Cardiorentis) to reflect increased costs anticipated by reason of Protocol Amendment 1 (explained below).
36. Change Order 2 was signed by the parties on 13 March 2014. It provided for an increase of about Eur 3.07 million in IQVIA’s budgeted own costs and of c. Eur 1.48 million in budgeted pass-through costs to reflect increased costs anticipated by reason of Protocol Amendments 1 and 2. This Change Order also provided for a revised payment schedule.
37. Change Order 5 had an effective date of 19 December 2014. It provided for further changes to the budget to reflect an extension of the anticipated timeline of the study in light of observed recruitment rates, as well as changes necessitated by Protocol Amendment 3. Overall, it provided for an anticipated increase in IQVIA’s own costs of c. Eur 7.06 million, and of pass-through costs of c. Eur 3.83 million.
38. Change Order 6 was signed by Cardiorentis and IQVIA on 30 June 2015. It provided for further changes to the budget to reflect another extension in the anticipated timeline of the study, as well as additional anticipated costs in relation to the addition of new countries to the geographical scope of the study. Overall, Change Order 6 provided for an anticipated increase in IQVIA’s own costs of c. Eur 6.63 million and of pass-through costs of c. Eur 4.62 million.
39. Change Order 7 has an effective date said to be ‘upon signature’. It was signed by Cardiorentis on 31 March 2016. Under it, the Service Delivery Excellence Program Milestones which had been agreed under the GSA were amended. In particular the two Performance Targets under the GSA relating to database lock - one requiring IQVIA to achieve database lock four weeks after ‘Last Patient out’ and the other requiring IQVIA to achieve database lock by 28 December 2014 - were removed and replaced with a new Performance Target requiring IQVIA to achieve ‘90% Data Clean’ by 29 February 2016. Other changes were also made to the budget and a revised project budget was enclosed indicating Total Study Fees of Eur 80,326,108.89. Change Order 7 provided for a revised payment schedule for IQVIA’s own fees, with pass-through costs continuing to be invoiced monthly as incurred.
The Protocol
40. Key to the conduct of a clinical study such as TRUE-AHF is the trial or study protocol. ICH GCP defines the trial protocol as ‘A document that describes the objective(s), design, methodology, statistical considerations, and organization of a trial.’ ICH GCP states that ‘The protocol usually also gives the background and rationale for the trial, but these could be provided in other protocol referenced documents.’ (ICH GCP 1.44). Section 6 of ICH GCP describes the topics which should generally be included in a trial protocol. These include (inter alia):
· A detailed description of the objectives and the purpose of the trial. (para 6.3)
· A description of the trial design, on which ICH GCP states that ‘[t]he scientific integrity of the trial and the credibility of the data from the trial depend substantially’. (para 6.4)
· The subject inclusion and exclusion criteria. (para 6.5)
· The specification of the efficacy parameters, and the methods and timing for assessing, recording and analysing these efficacy parameters. (para 6.7)
· The number of subjects planned to be enrolled, together with the reason for the choice of the given sample size, including reflections on or calculations of the power of the trial and clinical justification. (para 6.9)
· The level of statistical significance to be used. (para 6.9).
41. The first Clinical Study Protocol for TRUE-AHF was dated 19 April 2012.
42. This original Study Protocol recorded the objective of TRUE-AHF as being ‘to evaluate the effect of a 48-h continuous IV [viz intravenous] infusion of ularitide (15 ng/kg/min) versus placebo on the clinical status of patients with ADHF’. The Protocol specified that TRUE-AHF was to be undertaken on a randomised, placebo-controlled, double-blind basis.
43. In this context ‘placebo-controlled’ meant that trial subjects were to be divided into two groups: the ‘treatment’ group, which would receive infusions of ularitide, and the ‘control’ group, which would receive matching infusions of a placebo substance. ‘Randomisation’ is the process by which trial subjects are assigned either to the treatment or the control group, using an element of chance to determine the assignments in order to reduce bias. For TRUE-AHF, the Protocol specified that assignments were to be on a 1:1 basis, meaning that half of the trial subjects would be enrolled into the treatment group, and the other half would be enrolled into the control group. ‘Double-blind’ meant that the trial subjects, the investigators, the monitors, the Sponsor and anyone else involved in the running of the study (save for the independent safety board) would all be unaware of trial subjects’ treatment assignments, again to reduce the possibility of bias.
44. To evaluate the effect of short-term ularitide infusions in patients, the original Protocol proposed to test a single ‘efficacy endpoint’, which was in the following terms:
‘Improvement in a hierarchical clinical composite comprised of elements associated with: patient global assessment using a 7-point scale of symptomatic improvement, lack of improvement, or worsening; persistent or worsening heart failure (HF) requiring an intervention (initiation or intensification of IV therapy, circulatory or ventilatory mechanical support, surgical intervention, ultrafiltration, hemofiltration or dialysis); and all-cause mortality. Assessment of the clinical composite will be performed at 6 hour (h), 24 h and 48 h after start of IV ularitide infusion. Patients will be classified as “improved” if the patients are moderately or markedly improved at all 3 time points (at 6h, 24 h and 48 h) and do not fulfil criteria for “worse” during the first 48 hours following the start of the study drug infusion. Patients will be classified as “worse” if (during the 48 h) they die; experience worsening HF requiring a prespecified intervention at any time during the first 48 h; or experienced moderate or marked worsening of their global assessment at any of the 3 time points (at 6 h, 24 h or 48 h).’
45. In broad terms, therefore, following a subject's enrolment into the trial, the subject would receive an intravenous infusion of ularitide (if they were in the treatment group) or of a placebo substance (if they were in the control group). At 6 hours, 24 hours and 48 hours after the start of this intravenous infusion, the subject would be asked to rate how they felt their condition had changed (in each case, assessed by reference to how they felt at the time the ularitide/placebo intravenous infusion was first started) using a 7-point scale as follows:
1. Markedly improved
2. Moderately improved
3. Slightly improved
4. No change in status
5. Slightly worse
6. Moderately worse
7. Markedly worse
46. Subjects would then be classified into three groups depending upon their responses to these three assessments and depending upon any events arising during the 48-hour period of the infusion. The classification would be as follows:
· If a subject died during the 48-hour period, they would be classified as ‘worse’.
· If a subject experienced persistent or worsening HF during the 48-hour period (as assessed by an independent adjudication board), they would also be classified as ‘worse’.
· Subject to not falling within either of these categories they would be classified depending upon their responses to the 7-point assessments as follows: (i) subjects who stated they felt moderately or markedly worse at any of those three assessments would be classified as ‘worse’; (ii) subjects who stated they felt moderately or markedly improved at each of those three assessments would be classified as ‘improved’; and (iii) all remaining subjects would be classified as ‘unchanged’.
47. Following this process, a comparison would be made between the performance of the treatment group and the performance of the control group (in terms of the number/proportion of those subjects in each group which were ‘improved’, ‘unchanged’ and ‘worse’ after the end of the 48-hour period) and an assessment would be made of the extent and statistical significance of any difference identified.
48. The original Protocol, in addition to the primary efficacy analysis described above, included a primary safety variable which was defined as ‘the proportion of patients that have died or had a cardiovascular rehospitalization up to Day 30’ and the hypothesis which was being tested was that ularitide would have a non-inferiority margin (as compared to the control group) of no more than 4%.
49. The Protocol also specified certain ‘Secondary Endpoints’, namely (i) changes of NT-pro BNP at 48 hours of treatment compared to baseline, (ii) all-cause mortality and cardiovascular rehospitalization at Day 90 after start of study drug infusion, and (iii) cardiovascular mortality at Day 90. In addition, the Protocol identified certain ‘Exploratory Endpoints’, and these included ‘combined risk of all-cause mortality or cardiovascular rehospitalization at Day 60 and Day 180 after start of study drug infusion.’
50. In relation to the necessary or desirable size of a study, ICH Guideline on Statistical principles for clinical trials (E9) provides (ICH E9):
‘The number of subjects in a clinical trial should always be large enough to provide a reliable answer to the questions addressed. This number is usually determined by the primary objective of the trial. If the sample size is determined on some other basis, then this should be made clear and justified. For example, a trial sized on the basis of safety questions or requirements... may need larger numbers of subjects than a trial sized on the basis of the primary efficacy question...
The method by which the sample size is calculated should be given in the protocol, together with the estimates of any quantities used in the calculations... In confirmatory trials, assumptions should normally be based on published data or on the results of earlier trials. The treatment difference to be detected may be based on a judgement concerning the minimal effect which has clinical relevance in the management of patients or on a judgement concerning the anticipated effect of the new treatment, where this is larger...’
51. Consistently with this, the original Protocol stated in relation to the efficacy endpoint that:
‘Based on previous studies of ularitide, 46.9% of the patients receiving ularitide 15 ng/kg/min and 22.4% of the patients receiving placebo were successful in achieving marked or moderate improvement of dyspnea at 6 h which was still apparent at 24 h.
In the proposed Phase III trial, the evaluation of the composite endpoint will compare the distributions of patients who have improved, are unchanged, or who have worsened. It will be assumed that this distribution in the placebo group will be 25% improved, 50% unchanged, and 25% worsened. It is expected that use of ularitide will result in a relative increase of 50% in the percentage of patients who have improved, and a relative decrease of 33% in the percentage of patients who have worsened. As a result, the expected distribution of responses in the ularitide group is 37.5% improved, 45.8% unchanged, and 16.7% worsened.’
52. The Protocol provided that based on these assumptions, and taking account of the specified 2-sided p-value of 0.01, the trial would require 400 evaluable patients per treatment group, thus a total of 800 evaluable patients were required for the efficacy endpoint.
53. However, for the primary safety endpoint, the Protocol stated that:
‘The primary safety endpoint will evaluate the non-inferiority of ularitide to placebo in the rate of death or first rehospitalization for cardiovascular reasons at Day 30. Based on prior published data, the 30-day safety event rate in the placebo group is estimated to be 16%. The safety event rate of 14.9% is assumed in the ularitide group (corresponding to observed risk ratio of 0.93 from ASCEND-HF trial). Demonstrating the non-inferiority of the ularitide safety event rate to placebo within a non-inferiority margin of 4% using an exact binomial test (overall Type I error = 0.025, 1-sided; 90% power; 1:1 randomization) will require 1,058 evaluable patients per treatment group.’
54. Thus, in order to be able to demonstrate, to the specified level, the non-inferiority of ularitide in respect of the primary safety endpoint, more patients were required than to evaluate the efficacy endpoint, and this larger sample size was selected for the TRUE-AHF trial. The original Protocol stated that it intended to recruit some 2,116 patients.
55. The original Protocol set out, in Section 3, the Inclusion and Exclusion Criteria for patients to be eligible to be part of the study. To be eligible patients must have met all the inclusion criteria and must not have met any of the exclusion criteria. The original Protocol specified seven inclusion criteria and 21 exclusion criteria.
56. Inclusion criteria (or ‘IC’) 1, 2 and 3 were as follows:
‘1) Males and females aged 18 to 85 years
2) Unplanned hospitalization or emergency department visit for ADHF. Acute HF is defined as including all of the following:
a) Dyspnea at rest in a recumbent position (30 to 45 degrees), which has worsened within the past week.
b) Radiological evidence of HF on a chest X-ray.
c) BNP >500 pg/mL or NT-pro BNP >2000 pg/mL
3) Ability to start infusion of the study drug within 12h after initial clinical assessment performed by a physician at the emergency room/hospital with symptoms of ADHF.’
Inclusion criterion 3 reflected the aim of the study to have the commencement of infusion at as early a stage as possible after presentation.
57. Amongst the other inclusion criteria was inclusion criterion 6 (or ‘IC6’) which appeared in the original Protocol and versions 01 and 02 of the Protocol in these terms:
‘6) Persisting dyspnea at rest despite standard background therapy for ADHF (as determined by the Investigator) which must include IV furosemide (or equivalent diuretic) at ≥40 mg (or its equivalent) at any time after start of emergency services (ambulance, emergency department, or hospital). At the time of randomization, the patient must still be symptomatic. In addition, the patient should not have received an IV bolus of a diuretic for at least 2 h prior to randomization, and the infusion rates of all ongoing IV infusions must not have been increased or decreased for at least 2 h prior to randomization.’
In version 03, dated 8 December 2014, the last sentence was amended to insert the words ‘of medication to treat HF’ after ‘IV infusions’.
58. Also of significance is exclusion criterion 3 (or ‘EC3’). That criterion was specified in the original, and subsequent versions of, the Protocol as:
‘3) Treatment with levosimendan, milrinone, or any other phosphodiesterase inhibitor within 7 days before randomization.’
59. The original Protocol further set out in detail, in Section 4, the study treatment, and the methodology for storage, preparation, infusion, assignment and dosing of the study treatment(s).
60. In Section 6, the study schedule was set out. This stated that all timepoints were by reference to the start of study drug infusion (which was called t0). The schedule for treatment and for evaluations, including post treatment up to Day 180, was set out.
61. Section 7 of the Protocol set out the Statistical plan. Section 7.1.2 of the original Protocol provided:
‘The Full Analysis set (also ITT [ie Intention to Treat]) will be the primary analysis population. The primary efficacy analysis will be carried out on the 2-sided 0.99% level. The primary safety analysis will be carried out on the 1-sided 2.45% level. Nominal p-values will be presented for additional supportive analysis. Statistical tests on the primary efficacy endpoint will be repeated on the Per Protocol (PP) population to check the robustness of the test results.’
62. Section 7.1.2.1 explained and defined various terms used in Section 7.1.2, as follows:
‘There will be 3 analysis populations defined for this study:
· The Full analysis set (FAS; ITT) will be defined as all randomized patients, according to the ITT principle.
· The PP analysis set will be defined as patients in the FAS who do not have any major protocol deviations during the study treatment. Protocol deviations will be identified and documented following a blind data review prior to database lock.
· The As Treated (AT) set will be defined as all patients who received the study drug (active or placebo) classified according to the treatment that they actually received.’
63. Section 7 of the Protocol also provided that:
‘Details of imputations for missing data will be presented in the Statistical Analysis Plan (SAP), which will be finalized prior to database lock and study unblinding.’
64. The Protocol referred to the establishment and roles of two specific committees.
(1) An independent Clinical Events Committee (or CEC) which would adjudicate all efficacy and safety outcomes associated with the primary endpoints, and on all cardiovascular hospitalizations and deaths recorded during the 180 day follow up period.
(2) An independent Data and Safety Monitoring Board (or DSMB) which was set up to monitor safety aspects of the study and perform a review of progress towards primary study objectives.
65. Section 8 of the Protocol defined Adverse Events (‘AEs’) and Serious Adverse Events (‘SAEs’), and set out the requirements for documenting, assessing and reporting AEs occurring during the study period.
66. Section 10 of the Protocol provided that the Protocol, patient information, the Informed Consent Form (or ‘ICF’), and the Investigator’s Brochure should be submitted to the Institutional Review Board (‘IRB’) or an Independent Ethics Committee (‘IEC’) of the relevant sites and to the country regulatory authority (where applicable) for review and approval before the start of the study. The IRB/IEC and the country regulatory authority (where applicable) were to be informed of all subsequent protocol amendments and of SAEs occurring during the study.
Communications with the FDA in 2012
67. A full set of the communications of Cardiorentis with the FDA about the study was not before the court. The documents which there were showed that CardioPep and subsequently Cardiorentis had been in engaged in discussions with the FDA in relation to a new drug application for ularitide since at least 2010. A meeting was held between Cardiorentis and the FDA on 1 May 2012, at which the proposed Protocol for the study was submitted to the FDA. Presumably this was the original Protocol, to which reference has been made above. A slightly amended version of the Protocol, which included a population pharmacokinetic analysis plan, was submitted by Cardiorentis to the FDA on 23 July 2012.
68. On 10 August 2012 the FDA wrote to Cardiorentis stating that it had completed its review of Cardiorentis’s submissions and had a number of comments and requests. The FDA questioned the hierarchical clinical composite. One of the points made was:
‘If your proposed primary efficacy endpoint has favorable effects on global clinical assessment only and no adverse effects on mortality, a single trial with a highly significant p-value, as you are currently planning, would be required for approval of ularitide. Lacking this, a second trial would be required. Also, if the effect is all on the global assessment, you may not have an adequately reassuring safety database.’
69. On 24 September 2012 the multinational pharmaceutical company Novartis issued a press release with the headline results of a study, called RELAX-AHF, which it had been conducting into the efficacy of serelaxin, a drug with similarities to ularitide, in the treatment of ADHF. The title of the press release stated: ‘Results from Novartis Phase III study show that RLX030 [seralaxin] reduced deaths in patients with acute heart failure’. The first paragraph of the press release stated:
‘Phase III study results show that investigational RLX030 (seralaxin) reduced all-cause mortality in patients with acute heart failure (AHF). The six-month RELAX-AHF study shows that RLX030 reduces the number of deaths in patients with this disease, which has a higher mortality rate than most other cardiovascular diseases.’
70. In light of these results, Cardiorentis considered that it needed to investigate whether ularitide had an effect on mortality. Dr Holzmeister said, and I accept, that this was for three reasons: (1) first, that there was an ethical obligation to do so; (2) second, that if Cardiorentis did not do so and if Novartis succeeded in demonstrating a positive effect of seralaxin on mortality, Novartis would have a more valuable and marketable product than Cardiorentis; and (3) if Cardiorentis could show that ularitide did have a positive impact on patient mortality, then it would have a more valuable and marketable product. Cardiorentis accordingly proposed to the FDA the addition of cardiovascular mortality as a new co-primary endpoint for the TRUE-AHF trial. A meeting was held with the FDA on 6 December 2012. The minutes of that meeting record the following areas of agreement:
‘- The original clinical composite primary endpoint (consisting of use of 7-point patient global assessment of symptomatic improvement, assessment of patient improvement/worsening, and death) would continue to be evaluated for superiority to placebo with an assigned alpha of 0.01.
- A new cardiovascular mortality (using a time-to-event analysis) would be evaluated for superiority to placebo with an alpha of 0.04. To evaluate this endpoint, the trial would continue until a total of 655 cardiovascular deaths had occurred.’
71. As further explained in the minutes, to test the new co-primary endpoint of cardiovascular mortality required a sample size of 2,152 randomized subjects. This would accordingly necessitate the addition of some 36 patients to the number it had hitherto been planned to recruit.
The amendment to the Protocol
72. To embody the decision to add a new co-primary efficacy endpoint, the study Protocol was amended. This amendment was finalised on 7 February 2013, and was called Protocol Version 01. Under this, it was specified that freedom from cardiovascular mortality during follow up after randomisation for the entire duration of the trial was a co-primary endpoint. This has been referred to during the hearing as ‘Co-primary Endpoint 2’, or ‘Endpoint 2’, and the original efficacy endpoint as ‘Co-primary Endpoint 1’ or ‘Endpoint 1’. The amended Protocol also made a modification to exclusion criterion 5 and added two new exclusion criteria.
73. The amended Protocol, in its statistical analysis plan, provided for some changes to the test to be used for the efficacy analyses in respect of Co-primary Endpoint 1, as well as providing for efficacy analyses in respect of Co-primary Endpoint 2, in the following terms:
‘Primary Efficacy Analyses
The following hypotheses will be tested:
Co-primary efficacy endpoint 1
H0: There is no difference in the distribution of hierarchical composite variable in the 2 treatment groups i.e., ularitide and placebo
The alternative hypothesis will be 2-sided and is stated as:
H1: There is a difference in the distribution of hierarchical composite variable in the 2 treatment groups.
The primary efficacy analysis for co-primary endpoint 1 will be performed on the FAS. The composite primary efficacy endpoint will be tested using the Cochran-Mantel-Haenszel test for singly ordered data (with time from first physician evaluation to the start of study drug infusion [≤6h vs >6h] and baseline SBP [≤ median vs.>median] as stratification variables) to compare results between treatment groups. A final p-value of 0.01 or less (2-sided) will be considered evidence of statistically significant superiority.
Co-primary efficacy endpoint 2
H0: There is no difference in the cardiovascular mortality between the 2 treatment groups
The alternative hypothesis will be 2-sided and is stated as:
H1: There is a difference in the cardiovascular mortality between the 2 treatment groups.
The primary efficacy analysis for co-primary efficacy endpoint 2 will be a time-to-event analysis performed on the FAS. The endpoint will be tested using a Cox proportional hazards regression analysis to compare results between treatment groups, with age, gender, SBP at baseline, the time (in hours) from the first physician evaluation to start of study drug infusion (≤6 h vs. >6 h), and region (North America, Latin America, Europe) included as covariates. A final p-value < 0.04 (2-sided) for the coefficient associated with study group — adjusted for interim analyses according to a Lan-DeMets spending function, as described in the DSMB charter and the SAP — for treatment group will be considered evidence of statistically significant superiority.’
74. In respect of sample size calculations following from this amendment to the Protocol, there was no change to the number of patients required to evaluate Co-primary Endpoint 1, ie 400 evaluable patients per treatment group. However, in relation to Co-primary Endpoint 2, the amended Protocol stated:
‘Co-primary efficacy endpoint 2: cardiovascular mortality
Cardiovascular mortality will be evaluated in a time-to-event Cox proportional hazards regression analysis. It is assumed that the risk of cardiovascular mortality will be 11.0% per 6-month period of follow-up in the placebo group and that this risk will be reduced by a relative 22% in the ularitide group to a rate of 8.58% per 6 months. Based on these assumptions, an overall Type I error level = 0.04 (2-sided), 90% power, and a Kaplan-Meier log-rank analysis to estimate sample size, a total of 682 cardiovascular deaths would be required to demonstrate superiority of ularitide. If enrollment takes place over a 2-year period and there is a minimum follow-up of 1 year, a total enrollment of 2,152 subjects is estimated to be required.’
75. The amended Protocol also provided for an interim analysis to be performed by the DSMB to assess whether the trial had any realistic prospect of yielding a positive outcome in respect of Co-primary Endpoint 2. The provision for this in the amended Protocol was as follows:
‘...under the direction of the DSMB, an analysis will be carried out to estimate conditional power when 50% of the number of cardiovascular deaths needed for evaluation of primary efficacy endpoint 2 (approximately 341 deaths) has been documented, or alternatively, when enrollment is expected to be completed within 60 days, whichever occurs first. The interim analysis will be based on only data collected for co-primary efficacy endpoint 2 and will be used to estimate the conditional power to achieve the original primary study objective and to potentially adaptively re-estimate the sample size requirement for this endpoint.
The specific details regarding the DSMB organization and procedures will be outlined in the DSMB Charter and a separate SAP will be produced covering the analysis required.’
76. During July to at least October 2012 there were discussions between Cardiorentis and IQVIA as to whether IQVIA should provide a quality assurance (or ‘QA’) audit service in respect of the TRUE-AHF trial. IQVIA put together a costed Quality Assurance Audit Proposal in October 2012. Cardiorentis ultimately opted not to use IQVIA to conduct its planned audits.
77. At least in part as an alternative to such an arrangement with IQVIA, Cardiorentis entered into a number of agreements with other organisations to provide aspects of quality assurance. These were:
(1) International Medical Research Consulting (also referred to in some documents as IMR Partner GmbH) (‘IMR Consulting’), which was a company which provided services in clinical research, including but not limited to medical monitoring, quality assurance, pharmacovigilance, regulatory affairs and medical writing. IMR Consulting and Cardiorentis entered into a Master Service Agreement on 4 February 2013 for the provision of certain specified services for the TRUE-AHF trial. IMR Consulting's role and responsibilities were set out in a series of six work orders which spanned the period February 2013 to August 2014. IMR Consulting's scope of services included:
· ‘Study oversight, incl. document review of study plans, reports, protocols, submissions, correspondence;
· Remote medical monitoring, including assessing the patient data in the eCRF [viz the electronic Case Report Form, explained below];
· Review of reports extracted from CTMS [viz the Clinical Trial Management System, explained below] or Infosario, e.g. site visit reports, SAE reports;
· Co-monitoring on-site, including review of site visit reports and monitoring guidelines
· Monitoring audits
· Sponsor representation in meetings, calls (QA and operational team)’.
(2) Tachris AG, which was a company which provided services in quality assurance including, but not limited to, investigator site audits, clinical trial related audits, vendor and other third party audits, quality and management system evaluation and development. On about 9 April 2013, Cardiorentis and Tachris AG entered into a Master Services Agreement for the provision of certain specified services for the TRUE-AHF trial. Tachris AG conducted audits at four investigator sites on behalf of Cardiorentis.
(3) Cardiorentis engaged two consultants to assist it with the TRUE-AHF trial: Dr Jürg Lustenberger of SwissPharmAudit GmbH, was engaged as Head of Quality Assurance and to assist Cardiorentis set up a quality management system in October 2013; and Dr Richard Holcomb as the Sponsor's biostatistician for the trial.
(4) Cardiorentis also contracted auditors from SwissPharmAudit GmbH (Anke Zampich and Michelle Sceppa) and from Adamas Consulting (Carly Davenport and Lizbeth Elliott) to assist it with, and conduct on its behalf, CRO systems audits and GCP investigational site audits during the course of the TRUE-AHF trial.
78. In addition Cardiorentis made a specific agreement with an IQVIA entity designed to ensure that effective communication and escalation procedures were in place between Cardiorentis and IQVIA’s own quality assurance function. This was the Clinical Quality Agreement (‘CQA’), entered into between Cardiorentis and IQVIA RDS Inc on 23 April 2013.
79. Clauses 1 and 2 of the CQA provided:
‘1. Purpose
This Quality Agreement defines and describes the general responsibilities and working relationships between Quintiles, Inc.’s Clinical Quality Assurance (Quintiles QA) function and Cardiorentis Limited (Cardiorentis) with respect to the Clinical Services Quintiles will provide for Cardiorentis clinical trials.
The purpose of this Quality Agreement is to confirm that effective communication and escalation processes are in place to allow optimal and immediate sharing of information between Quintiles QA and Cardiorentis. This Quality Agreement is in addition to any project specific communication plans in effect at this time as well as the General Services Agreement between Cardiorentis and Quintiles (dated 30 August 2012). In the event of a conflict between the existing agreements, the General Services Agreement will control.
2. Scope
This Quality Agreement applies to all the QA processes and procedures for the Cardiorentis QA and Quintiles QA staff and all staff working on Cardiorentis ULA01 study.”
80. Clause 4.4.1 of the CQA provided:
‘Project Quality Issues at IQVIA's monitored sites / facilities or processes associated with the TRUE-AHF trial would be processed by IQVIA's staff assigned to the TRUE-AHF trial in accordance with SOP CS_OP_QA002: Managing Quality Issues including suspected misconduct. Confirmed quality issues were to be escalated to the IQVIA Global QA Lead, who would escalate confirmed critical quality issues to the assigned Cardiorentis QA representative according to SOP CS_OP_QA002.’
81. Clause 4.4.2 of the CQA provided:
‘Confirmed Critical Quality Issues will be notified to Cardiorentis QA within one business day of being confirmed as critical by IQVIA QA and major quality issues would be notified to Cardiorentis within seven business days of IQVIA being notified. Both IQVIA QA and Cardiorentis would work together to resolve quality issues, determine the root cause and generate an effective CAPA [Corrective and Preventive Action] plan.’
82. Clause 5.2 of the CQA required non-audit activities between IQVIA QA and Cardiorentis would include, but not be limited to:
(a) “Monthly” QA to QA meetings/teleconferences dealing with Quality Issues, audits and audit CAPAs and any project level questions or concerns escalated to IQVIA QA by the IQVIA staff or other agents of Cardiorentis (as listed in Appendix 1 of the CQA).
(b) Participation of the IQVIA Global QA Lead or designee and Cardiorentis representatives (as listed in Appendix 1 of the CQA) in teleconferences to discuss relevant items related to the TRUE-AHF trial.
(c) Risk management of existing processes and introduction of process improvements, as applicable to the TRUE-AHF trial.
(d) Regulatory Authority Inspection and customer audit support (if required)
(e) Providing QA and compliance guidance to the project team
(f) Quality and Compliance training, including lessons learned (where applicable).’
83. The TRUE-AHF trial involved a considerable number of third parties, some of whose roles it is necessary to record before considering the progress and conduct of the trial.
Investigators
84. ‘Investigators’ were the medical professionals who were to conduct the clinical trial at the relevant ‘sites’. The sites were the hospitals and medical centres in each country where the TRUE-AHF trial was conducted. Neither the investigators nor the other staff of the sites were employees of either Cardiorentis or of IQVIA.
85. ICH GCP E6 (R1) (para. 4.1.1 - 4.1.2) provided that investigators should be qualified by education, training and experience to assume responsibility for the proper conduct of the trial, and should be ‘thoroughly familiar with the appropriate use of the investigational product(s), as described in the protocol, in the current Investigator's Brochure, in the product information and in other information sources provided by the Sponsor.’
86. It was the investigators who were responsible for verifying that each patient enrolled met the eligibility criteria for the study at the time of enrolment.
87. Section 10.4 of the Protocol for the TRUE-AHF study (in its original and amended forms) required investigators to apply due diligence to avoid protocol deviations.
88. Under ICH GCP E6 (R1) para 4.1.5 investigators were permitted to delegate significant trial-related duties to appropriately qualified persons (but were required to maintain a list of those persons). Principal Investigators (or ‘PIs’) were the investigators with ultimate responsibility for the conduct of the clinical trial at their particular site. In addition, some sites had sub-investigators, study nurses and other staff who would also be involved in patient care for subjects on the clinical trial.
The Executive Committee
89. An Executive Committee of 12 independent scientific advisors and investigators and four non-voting Cardiorentis team members (including the CEO, and Cardiorentis's biostatistician) was established for the TRUE-AHF trial. The Chairman and Principal Investigator of the TRUE-AHF Executive Committee was Dr Milton Packer. Additional Cardiorentis and IQVIA team personnel could also attend meetings as observers to provide status and operational updates on the study progress. The Executive Committee was responsible for the overall guidance of the study, would review and finalise the TRUE-AHF protocol and any amendments and oversee the conduct of the Clinical Events Committee and the DSMB.
The Steering Committee
90. A global Steering Committee was also established for the TRUE-AHF trial, made up of 29 investigators and Key Opinion Leaders (KOLs). KOLs were respected members of the medical/scientific community, who were chosen to be champions of the study. The Steering Committee was responsible for, amongst other things, identifying suitable sites in relevant countries, supporting sites with protocol training and questions on the protocol or study procedures, checking recruitment rates in relevant sites and developing recruitment strategies as needed, attending monthly KOLs Update meetings and chairing monthly ‘country update’ meetings. Meetings were held by European Steering Committee members and Global Steering Committee members periodically.
Documents detailing practices and procedures for the TRUE-AHF trial
91. During the course of the study, IQVIA, with some input from Cardiorentis and Dr Packer, produced a suite of documents to elaborate and set forth the practices and processes to be followed in relation to various aspects of the trial.
Monitoring Guideline
92. One of these documents was the Monitoring Guideline.
93. There were three versions of the Monitoring Guideline, but only two operative during the trial period, namely version 1 dated 1 February 2013 and version 2 dated 15 October 2015. The Monitoring Guideline stated that IQVIA was responsible for ‘all monitoring activities’ in all the countries involved in the trial, save for Israel.
94. The Monitoring Guideline set out the expected activities of the Clinical Research Associates (or ‘CRAs’). CRAs were to act as a liaison between the Sponsor or CRO and the trial site. Their role was to monitor compliance with the clinical trial protocol, check activities on site, review CRFs and perform various other functions. For all countries other than Israel, the CRAs for the trial were engaged by IQVIA.
95. Section 1.2 of the Monitoring Guideline provided that ‘the CRA will review all protocol deviations with the Investigator during monitoring visits to address corrective measures to be taken, identify actions required to prevent future protocol deviations and evaluate if this deviation could be a hazard to the patient’.
96. This section also contained timelines for reporting and review of protocol deviations as follows.
· ‘All protocol deviations identified in addition to the resulting actions should be documented in the monitoring visit report and the follow-up letter. Protocol deviation should be reported in CTMS within 10 days of the visit.’ [This was changed to 5 days in version 2 of the Monitoring Guideline].
· ‘CPMs’ [viz Clinical Project Managers, who manage the operational conduct of the study and oversee the coordination of the staff assigned to it] ‘and the Medical Advisor(s) will review listings of protocol deviations entered into CTMS, with a focus on critical PDs [viz Protocol Deviations], on a monthly basis during the active phase of the study to identify trends and to discuss potentially required preventive actions and/or corrective actions.’
· ‘The outcome of the meetings will be discussed with Cardiorentis and listings and trends of protocol deviations will be provided to Cardiorentis following the meeting.’
97. Section 1.4 provided:
‘Site training
Sites will be trained on the protocol procedures by the CRA during the Site Initiation Visit (SIV) and if necessary during subsequent Monitoring visits. The Quintiles CRA is responsible for ensuring that all site training is documented by the site personal (sic) in the site specific Training Log.’
98. Section 2.1 provided that the first monitoring visit should always occur within two weeks from first patient randomisation and the frequency of the Interim Monitoring Visits would be no greater than 8-10 weeks. Monitoring visit frequency would need to be increased depending on patient recruitment and volume of work. The site visit report was to be submitted in the five days following the visit. Protocol deviations were specified as one of 9 items that should be part of an ongoing review of reports and data.
99. In Section 5.2, under the heading, ‘SDV Requirements’, it was stated that ‘For this study, CRAs are asked to conduct 100% SDV for all patients consented to the study.’ The relevant description of SDV under section 5.2 notes: ‘CRA will be completing review of all source documents against data entered into the eCRF.’
100. Section 5.3 of the Monitoring Guideline, under the heading, ‘Data to be 100% verified’, provided, amongst other things:
(i) Under the sub-heading ‘SAEs/Endpoints’, ‘…For all SAEs/Endpoints that occur during the trial through time of site close out perform 100% SDV to ensure consistency between data in eCRF, SAE report, MERGE and source documents as per protocol requirements…’
(ii) Under the sub-heading ‘Inclusion/Exclusion criteria’, ‘All the eligibility criteria will need to be carefully verified vs. source documents. Patients should not be screened if adequate source documentation of eligibility is missing or incomplete…’
(iii) ‘All eCRF pages.’
(iv) Under the sub-heading, ‘Concomitant Medications’, ‘Sites to document and CRAs to review planned treatment for each subject at the time of randomization. CRA to review all concomitant medications and documentation indicating if these were given as standard treatment or if they are a result of worsening heart failure. If given for worsening heart failure, this should be reported as an SAE and Endpoint.’
101. Section 6.1 of the Monitoring Guideline, under the heading, ‘Specific Requirements for Data Capture’ stated ‘Data has to be entered in eCRF by the site within 3 working days after patient’s visit, or immediately after data becomes available to the site…’
102. This Plan was dated 3 May 2013. It specified the various roles of those involved in the study for IQVIA.
103. It further set out the intended procedures for ‘Site Compliance Issue Escalation’ and ‘Quality Issue Escalation’. As part of the former, it was specified that ‘if non-compliance issues or indicators of quality issues are observed, the CRA has to discuss the issue with the PI and relevant site staff either during a monitoring visit, where non-compliance was detected, or on the phone, if non-compliance was detected between scheduled monitoring visits’. It was further provided that: ‘The CRA shall involve the PI as an active participant in the discussion of site non-compliance and the plans for corrective action’; that ‘Once the site implements corrective action and it can be confirmed the correction addresses the issue then no further action should be required’, and the resolution should be documented in the site visit report; but that if the non-compliance continued and the CRA was unable to obtain satisfactory resolution of the issue with the PI, then the escalation procedure to the Regional CPM and Clinical Operations Quality Manager should begin.
104. This Plan also set out the intended procedures for Quality Management. These included a section on ‘Cardiorentis’ Oversight Responsibility’. It specified that ‘Cardiorentis and/or its representatives, e.g. contracted third party expert(s), will be working in close collaboration with Quintiles to ensure sufficient oversight over clinical study ULA01’. The oversight tools which would be used to control study performance included the following
|
Task |
Oversight Tools |
Quality Control Measure |
|
… |
|
|
|
Site Training |
Consistent global training with Cardiorentis’ involvement |
Approval of training material
Participation in training sessions to assess and provide continuous improvement |
|
CRA Training
… |
Consistent global training with Cardiorentis’ involvement |
Approval of training material
Participation in training sessions to assess and assessment of Quintiles staff participation (preparedness, focus of questions, etc)
|
|
Medical Monitoring |
Medical Monitoring Plan
Remote medical monitoring of eCRFs |
Review and approval of the plan
Comparative external review of medical information in the INFORM across patient, sites, countries, regions |
|
Protocol deviations |
Monthly Protocol Deviation Report (all sites) |
Review of report and deviation rate for trends and escalation with sites involving Key Opinion Leaders |
105. A further document in the suite of study documents produced by IQVIA was a Protocol Deviation Management Plan (or ‘PDMP’). The first version of this document was dated 21 January 2013, and there were subsequent versions dated 4 February 2013 and 12 December 2013. The third version contained the following:
‘ICH GCP section 5.20 states:
5.20.1 Non-compliance with the protocol, SOPs, GCP, and/or applicable regulatory requirements by an investigator/institution, or by member(s) of the Sponsor’s staff should lead to prompt action by the Sponsor to secure compliance.
5.20.2 If the monitoring and/or auditing identifies serious and/or persistent noncompliance on the part of an investigator/institution, the Sponsor should terminate the investigator’s/institution’s participation in the trial. When an investigator’s/institution’s participation is terminated because of non-compliance, the Sponsor should notify promptly the regulatory authority(ies).
‘ICF E3 (sic) section 10.2 Protocol Deviations (within the CSR) states: All important deviations related to study inclusion or exclusion criteria, conduct of the trial, patient management or patient assessment should be described. In the body of the text, protocol deviations should be appropriately summarized by center and grouped into different categories, such as:
− those who entered the study even though they did not satisfy the entry criteria;
− those who developed withdrawal criteria during the study but were not withdrawn;
− those who received the wrong treatment or incorrect dose;
− those who received an excluded concomitant treatment.
In appendix 16.2.2, individual patients with these protocol deviations should be listed, broken down by center for multicenter studies.’
106. The PDMP, in all its versions, identified fourteen types of protocol deviation (‘when an investigator site, or study subject, does not adhere to protocol stipulated requirements’). The PDMP explained that the CTMS would distinguish between 14 categories of protocol deviation, of which category 2 was ‘eligibility and entry criteria’, and that the protocol deviations ‘will be tracked’ using those categories. Where a deviation fell into more than one category, the ‘primary’ category was to be selected; but the PDMP stipulated that violations of eligibility or entry criteria (category 2) and violations of efficacy criteria (category 10) were to have priority, and that category 2 (eligibility and entry criteria) was ‘superior to’ category 10.
The PDMP explained category 2 as follows:
|
‘This code should be used to identify any deviations that involve a violation of an inclusion or exclusion criterion, and/or any subject screening procedures are missed or performed outside the specified time frame. |
Examples include:
Inclusion/exclusion criteria violated,
Study procedures performed outside the required timeframe
Use of an excluded medication during that screening or within the prohibited time period per exclusion criteria.
Not performing required laboratory assessments during screening/prior to enrolment into the study.’ |
107. There was also a code for violations consisting of the impermissible use of a concomitant medication (category 3); if the concomitant medication resulted in an inclusion or exclusion violation, then category 2 was to be used.
108. The PDMP provided that each deviation should also be categorised by its severity: ‘critical’, ‘major’, ‘minor’ or none. As with the categorisations, each deviation was to be placed in a single category (‘Do not duplicate deviation entry’).
109. The categorisations were defined as follows:
|
‘PD Severity |
Severity Definition |
Severity Example |
|
Critical |
Issues that threaten scientific, ethical, regulatory or business integrity and could invalidate the acceptability of a study (or part of it) to a Sponsor or regulatory body, or invoke regulatory action. Such issues require immediate attention and prompt action. |
Study procedures being performed on patients without consent.
Suspected fraud
Lack of appropriate blinding or routine bias that is against protocol instruction. |
|
Major |
Issues that impact scientific, ethical, regulatory or business integrity and which, if unattended[,] could become critical. Such issues require timely action. |
Repeated minor PD or PD that occurred repeatedly
Laboratory tests related to patient eligibility not available at the time of randomization |
|
Minor |
Issues could potentially indicate a systematic fault in the process, which could lead to a major or critical finding if repeated or escalated. Typically a ‘once [sic] off’ discrepancy, failure, or finding with no systematic pattern. |
|
|
None |
No impact on data integrity, patient safety or ethical conduct.’ |
|
110. The PDMP further contained the following:
‘Protocol deviations that occurred at the sites will be identified during Monitoring visits, in-house review and other site contacts. The CRA is responsible for discussing identified issues with the PI and the relevant site staff in a timely manner and to follow up on actions related to protocol deviations until resolution…
If possible, for each deviation the actual root cause that caused the event should be identified, by discussing with the site the actual reason for the PD occurrence. … Preventive actions to avoid the deviations happening again are also expected to be discussed and agreed with the PI.
The CRA identifying the deviation has to track all detected deviations in CTMS within 10 days of the identification and has to update CTMS entries with new information obtained as well as the closure date.
If there are critical PDs identified or site non-compliance suspected, the responsible CPM must be informed immediately via email or phone within 24 hours of identification.’
111. The PDMP also contained a process for the ‘Review of Protocol Deviation Listings’. This involved the CPMs and the Medical Advisors reviewing the PD listings entered into CTMS on a monthly basis. Once the CPMs had reviewed the listing, it was to be reviewed by the Medical Advisors and then sent to Cardiorentis for a PD Review call. There would then be follow up if needed. ‘… CPMs are responsible to implement corrective/preventive actions of trends identified during the review of PD listings, follow up on deviations that are still open and to communicate trends and preventive actions to the clinical team…’
The Medical Monitoring Plan
112. A further document of the suite was the Medical Monitoring Plan. Version 3 was dated 23 September 2013, version 4 was dated 10 March 2014, version 5 was dated 10 April 2014 and version 6 was dated 20 May 2014. All these versions provided at paragraph 3.1.3 that Quintiles’ Medical Advisors were to ‘assist CPMs in on-going protocol deviation review’ but that ‘Quintiles Medical Advisors are not contracted for the remote medical monitoring of eCRF data consistency.’
The Statistical Analysis Plan
113. Also part of the suite of documents was a Statistical Analysis Plan (or ‘SAP’). This was developed to describe the rules and conventions to be used in the presentation and analysis of efficacy and safety data for the study. The SAP described the data to be summarised and analysed including specific provisions as to the statistical analyses to be performed. A first version of the SAP was dated 25 September 2014, with version 2 dated 16 February 2015 and version 2.1 dated 16 March 2016.
114. Section 6.6 of the first version and version 2.0 of the SAP set out the Statistical Tests as follows:
‘The primary efficacy analysis for co-primary efficacy endpoint 1 will be carried out with a 2-sided 1% significance level. The final analysis for co-primary efficacy endpoint 2 will be carried out with a 2-sided 4% significance level. For all other analysis the default significant level will be (5%); confidence intervals will be 95% and all tests will be two-sided.’
115. This was not materially altered in version 2.1 though there was an additional provision as to the use of a 5% significance level for secondary efficacy endpoints.
116. In addition each version of the Statistical Analysis Plan contained provisions: in Section 7.3 dealing with Missing Data in general terms, in Section 7.4 dealing with Multiple Comparisons / Multiplicity, and in Section 7.5 dealing with Examination of Subgroups. Section 15.1.1 of each version dealt with Primary Efficacy Outcomes - Primary Efficacy Variables and Derivations. Section 15.1.2 laid down Missing Data Methods for Primary Efficacy Variables. Section 15.1.3 dealt with the Primary Analysis of Primary Efficacy Variables, and Section 15.1.4 dealt with Sensitivity Analysis of Primary Efficacy Variables.
117. According to the GSA, the trial was to be carried out at 194 sites in North America, Europe and Latin America, enrolling 2117 patients. In fact, at the point of last patient in, 150 sites had been initiated, and had enrolled and randomised at least one patient in the TRUE-AHF trial; and a total of 2157 patients had been enrolled and randomised.
118. The number of sites initiated and patients enrolled and randomised month by month is shown in the following table:
|
Month-end |
Total sites |
Total patients |
|
2012 |
Aug |
1 |
1 |
|
|
Sept |
1 |
1 |
|
|
Oct |
1 |
1 |
|
|
Nov |
2 |
2 |
|
|
Dec |
4 |
11 |
|
2013 |
Jan |
6 |
20 |
|
|
Feb |
6 |
28 |
|
|
Mar |
9 |
35 |
|
|
Apr |
22 |
71 |
|
|
May |
41 |
126 |
|
|
Jun |
62 |
193 |
|
|
Jul |
81 |
317 |
|
|
Aug |
89 |
416 |
|
|
Sept |
99 |
527 |
|
|
Oct |
104 |
655 |
|
|
Nov |
111 |
779 |
|
|
Dec |
118 |
871 |
|
2014 |
Jan |
123 |
973 |
|
|
Feb |
127 |
1066 |
|
|
Mar |
127 |
1159 |
|
|
Apr |
127 |
1235 |
|
|
May |
128 |
1297 |
|
|
Jun |
128 |
1374 |
|
|
Jul |
130 |
1445 |
|
|
Aug |
132 |
1503 |
|
|
Sept |
134 |
1579 |
|
|
Oct |
135 |
1654 |
|
|
Nov |
136 |
1726 |
|
|
Dec |
138 |
1780 |
|
2015 |
Jan |
140 |
1860 |
|
|
Feb |
141 |
1922 |
|
|
Mar |
144 |
2026 |
|
|
Apr |
149 |
2106 |
|
|
May |
150 |
2157 |
119. The first patient enrolled and randomised into the TRUE-AHF trial was patient 04090001 at site 0409: Ohio State University Medical Centre, USA on 15 August 2012. The last patient enrolled and randomised into the TRUE-AHF trial was patient 0403010 at site 0403: University of Cincinnati, USA on 17 May 2015.
120. At the beginning of the trial Mr Elmar Schnee was Cardiorentis’s CEO, and Dr Johannes Holzmeister was its CMO. Dr Richard Holcomb was involved in the project from early 2012 onwards as the Sponsor's Statistician. Dr Stefan Mazgareanu joined (initially seconded from CardioPep) in December 2012 (after a brief spell acting as a consultant from October 2012 onwards) as head of project management and regulatory affairs. In April 2013, Professor Markus Meyer (who, as I have already said, had a background of extensive previous involvement in ularitide) joined Cardiorentis and he became CMO in 2015 when Dr Holzmeister became CEO. Dr Henri van Langenberghe joined Cardiorentis in August 2013 as Director of Clinical Operations; Dr Teiba Al-Haboubi joined as Clinical Operations Manager in October 2013; and Mr Jürgen Rohner as Clinical Research Associate in March 2014. Mr Krzysztof Hoffman was also hired to assist Dr Mazgareanu in August 2013. Mr Reto Wittwer was hired as CFO in January 2015.
121. As for IQVIA:
(1) Dr Jeffrey Spaeder was, throughout the Trial, IQVIA’s Chief Medical and Scientific Officer. His first direct involvement in the trial was in 2013, and he became a high-level ‘point of contact’ with Cardiorentis’s senior management. Other senior management who seem to have had, principally, a client management role were Mr Hans van Dijk (Global Vice-President, Cardiovascular), and Mr Philip Galtry (Vice President, EMEA Operations Head). The operational management of the project was broadly hierarchically organised:
1. There was a ‘Senior Project Oversight’ role, which was initially filled by Mr Galtry (until September 2014), and thereafter by Mr Michael Abbs.
2. There was a ‘Project Lead’, whose role was primarily one of oversight. This was Ms Ana Gonzalez (until April 2013), Mr Matteo Mondellini (from April 2013), Mr Jason Turner (from the end of 2014), and then Ms Aline Ron (from early 2016). Ms Lara Queiroz thereafter took over the role of Project Lead as the Study was winding down.
3. CPMs took responsibility for the investigational sites in particular regions. Their role was to manage all aspects of IQVIA’s work in relation to clinical issues of a trial in their region, and in particular to manage the CRAs and the investigating sites within an assigned region. Ms Lara Queiroz, Ms Vaida Makunaite-Vilute, Mr Michal Meller and Ms Aline Ron were some of the IQVIA CPMs on the TRUE-AHF trial.
4. The task of communicating directly with Investigators and making the necessary site visits and so forth was that of CRAs.
(2) Alongside the management and clinical operational side, there was a separate quality assurance function. Again, this was hierarchically organised:
1. Mr Tony Owen was for most of the relevant period Global Head of Clinical Quality Assurance, and (from December 2015) Global Head of Quality Assurance. He had oversight of the provision of QA services in support of the complete portfolio of clinical trials managed by IQVIA, including the TRUE-AHF trial.
2. There was a designated Global QA Lead for the project. Over the course of the Study three people occupied that role: Ms Veronica Sandström (September 2012–October 2013), Ms Sherry Popiolek (October 2013 or thereabouts–August 2015), and Mr Kenny Van Speybroeck (from August 2015 onwards).
3. The QA Lead was assisted by others, such as Mr Satish Prabhu.
(3) There was, separately, a biostatistics (or BIOS) team, which was involved in developing statistical analysis of the Study’s results with Cardiorentis, and in particular with Dr Richard Holcomb. There was an ‘IQVIA statistical lead’, who was initially Mr Andrew Tomlinson and then, from late 2015, Ms Claire Raskino. Ms Raskino became the principal point of contact with Cardiorentis and BIOS Team Leader in October 2015.
(4) There was a Senior Medical Director, Dr Véronique Mahaux, and a Medical Director, Dr Rafal Ziecina, who provided medical input from time to time.
122. In the TRUE-AHF trial, patient data were to be collected in a centralised electronic database managed by IQVIA (the electronic data capture (eDC) system). This database was given the name ‘InForm’. Cardiorentis had (read-only) access to that database.
123. To facilitate the collection of these data, IQVIA created a trial-specific electronic Case Report Form (eCRF), which it provided to the sites. Authorised personnel at the sites (under the supervision and responsibility of the sites’ Principal Investigators) were then responsible for completing the eCRF for each patient.
124. Site personnel were responsible for ensuring that the information on the eCRF was comprehensive and matched the patients’ original medical records (as kept by the sites themselves). Where IQVIA had queries in respect of the information provided by the sites in the eCRF, IQVIA could raise queries on the data in the system, which site personnel were then required to review and answer as necessary.
125. Following the entry of all data for the trial by a site, the source document verification, or SDV, of that data by IQVIA and the resolution by the sites of all data queries raised by IQVIA, the site’s data for the trial would be locked and would no longer be editable.
126. This process is summarised in the Protocol, which provided that:
‘The eCRF and the protocol are both confidential. The eCRF will be created by the CRO and programmed into the electronic data capture (eDC) system. All study centers will need internet access to access the eCRFs and will only have access to data for patients at their own study centers. Data management (DM) and other coordinator teams will have access to data at all study centers.
All eCRFs are to be completed by an authorized member of the investigational staff and reviewed and signed by the Investigator. All entries, corrections, and alterations are to be made by the responsible Investigator or an authorized member of the investigational staff.
All eCRFs are to be completed in a manner that ensures accurate interpretation of data within 72 h of each study visit.
It is each Investigator's responsibility to ensure that all discontinued orders or changes in the study or other medications entered on the patient's eCRF correspond to the entries on the patient's medical records.
The eCRFs for any patient leaving the study should be completed at the time medication is terminated for whatever reason.
The eCRFs must accurately reflect data contained in patient's records (e.g., source documents).
After data is entered into the eCRF by the site, queries that are generated by the eDC system should be addressed by the site. Data queries will be raised for inconsistent, impossible, or missing data. All entries to the study database will be available in an audit trail.
Data Management or other parties involved can also raise manual queries for sites to address (e.g., for coding queries). The same process is to be followed by any other groups creating manual queries in the eDC system (e.g., for SAE reconciliation). Once all data is entered, source document verification (SDV) completed on required fields, manual queries and electronic data reconciliation completed and all queries closed, the casebook can be signed by the Principal Investigator. Once the casebook is signed, DM will then lock the casebook so that no modifications can be made.’
127. The eCRF itself was divided into a number of forms dealing with different aspects of the patient data. Guidelines were provided to the sites to assist them in completing these various forms.
128. First and foremost, the sites had to obtain subjects’ informed consent (including re-obtaining informed consent for those subjects enrolled prior to the first Protocol amendment) and this had to be documented in the ‘IC’ and ‘IC-AMEND1’ forms.
129. Sites then had to document subjects’ compliance with inclusion and exclusion criteria in the inclusion/exclusion criteria ‘IE’ (‘IEPA’ after Protocol amendments) form. That form specifically required sites to confirm the relevant patient’s compliance with each inclusion criterion and each exclusion criterion individually (with ‘Yes’ or ‘No’ answers in each case).
130. Sites then had to complete a number of other forms relating to screening, such as the medical history form (‘MEDHIS’), the vital signs and physical examination form (‘PE’), the chest X-Ray form (‘CXRAY’), and the hematology (‘HEMA’) form.
131. As a final step before randomisation of patients, the sites had to complete the ‘end of screening’ form (‘EOSCR’). This required the sites to re-confirm:
a. Whether the subject received IV furosemide (or equivalent) at ≥40 mg (or its equivalent) at any time after start of emergency services. The EOSCR form provided that if the answer to this question was ‘Yes’, sites should ‘ensure that the details are recorded in the Previous & Concomitant Medications form (CONMED)’. In this respect, the eCRF guidelines provided to the sites also stated that ‘If question 1 “Did the subject receive IV furosemide (or equivalent)…? Is answered YES, do not forget to record IV furosemide (or its equivalent) as part of the concomitant medications on the CONMED form’.
b. Whether the subject still had dyspnoea at rest in recumbent sitting position (30 to 45 degrees).
c. Whether the subject was receiving or had received (inter alia) levosimendan, milrinone, or any other phosphodiesterase inhibitor within 7 days before randomisation.
d. Whether, within the last 2 hours, there had been any change in the rate of infusion of a diuretic, vasodilator or inotropic drug.
e. Whether, within the last 2 hours, the subject had received a bolus of diuretic.
132. As noted above, and as set out in the eCRF guidelines which they were provided with, sites were required to enter on the previous and concomitant medication (‘CONMED’) form ‘all the medications taken within 7 days prior to the first administration of study drugs and during the trial’. Each medication administered was required to be entered into a separate entry recording the drug name, dose, route and frequency, the date and time the treatment was started and the date when the treatment was stopped (if applicable), and the reason for the administration of the medication.
133. Other forms also had to be completed to record the administration of the study drug (DRUGADMIN form), and to record the patient’s global assessment at the relevant time points (PGA form), as well as in the event of adverse events occurring (AE form), or death (DEATH form).
134. Sites were of course permitted to, and did, maintain their own patient records in addition to completing the eCRF forms. To assist them with the collection of the required trial data, sites were also provided by IQVIA with data collection worksheets (which it was common ground some sites used, but not others). The introduction to the data collection worksheets explained that:
‘These worksheets will be used to document important data points collected during the study. In some cases required information might be documented in other locations, for example, the subject medical record and do not need to be copied into these worksheets if they are known to be available elsewhere.
It is important to collect all necessary data at the time-point specified in the protocol and flowchart. All source documentation needed to determine eligibility for enrolment and for collection of data after start of study drug infusion should be completed in “Real Time”; do not complete them retrospectively.’ (emphasis in original)
135. The eCRF completion guidelines also provided (as did the Protocol) that all eCRFs were ‘to be completed in a manner that ensures accurate interpretation of data and further remote monitoring for safety reasons, within 72 h of each study visit’. The guidelines also provided that:
‘A query must be answered within 3 working days of receipt. 3 weeks prior to database lock this timeline is reduced to 24h. During the maintenance phase of the study please check the eCRFs at least twice a week for any newly issued queries to meet the timeline. Your CRA will review query resolution on an ongoing basis and will contact you should your site continually not respond to queries within 3 days of receipt. Metrics for query turnaround times are closely monitored.’
136. In the original GSA, IQVIA had anticipated that a total of 204,926 eCRF pages would be created for the TRUE-AHF trial. By 7 March 2016, IQVIA was recording that 333,976 eCRF forms had been completed.
137. Distinct from the eCRF database, IQVIA and Cardiorentis were required by the ICH GCP Guideline to maintain an electronic trial master file (or ‘eTMF’), the purpose of which was to preserve ‘essential’ study documents, being ‘those documents which individually and collectively permit evaluation of the conduct of a trial and the quality of the data produced.’
138. The ICH GCP Guideline provides a minimum list of essential documents which should be preserved in the eTMF, which includes among other things the study protocol, all written information given to the trial subjects, all signed agreements between the Sponsor, CRO and investigators, as well as monitoring visit reports, communications between sites and the CRO/Sponsor, and the case report forms.
139. Separate again was the Clinical Trial Management System (or ‘CTMS’). This was an internal IQVIA management system and only IQVIA personnel had direct access to it. It was used, inter alia, by CRAs to generate site monitoring visit reports, and to track action items and protocol deviations.
140. Protocol Deviation Trend Reports (or PD Logs) were prepared by IQVIA to tabulate the information as to protocol deviations identified by CRAs and recorded as such on the CTMS.
141. The first PD Log was sent to Cardiorentis on 4 February 2013. The next PD Log dated 13 March 2013 was sent to Cardiorentis on 21 March 2013. The third PD Log for 2013 was dated 28 May 2013 and sent to Cardiorentis on 9 June 2013.
142. The next PD Log (dated 9 October 2013) was sent to Cardiorentis on 4 November 2013, showing 839 violations of which 20 were of eligibility criteria. Two further PD Logs were issued in 2013: one dated 29 November 2013, sent on 10 December 2013; the other dated 27 December 2013 sent on 13 January 2014.
143. In 2014, 9 PD Logs were issued as set out below:
|
No. |
Date of the PD Log |
Date circulated to Cardiorentis |
|
|
31 January 2014 |
12 February 2014 |
|
|
28 February 2014 |
3 April 2014 |
|
|
28 March 2014 |
17 April 2015 |
|
|
3 June 2014 |
24 June 2014 |
|
|
8 July 2014 |
25 July 2014 |
|
|
4 September 2014 |
25 September 2014 |
|
|
2 October 2014 |
27 October 2014 |
|
|
31 October 2014 |
25 November 2014 |
|
|
November 2014 |
1 December 2014 |
144. In 2015, 10 PD Logs were issued and circulated to Cardiorentis on the following dates: 15 January 2015, 13 February 2015, 11 March 2015, 27 April 2015, 27 May 2015, 9 July 2015, 7 August 2015, 16 September 2015, 28 October 2015 and 10 December 2015.
145. There were two PD Logs issued in 2016: 15 January 2016 and 24 February 2016. The breakdown page on the February 2016 report showed a total of 6208 protocol violations, of which 175 were violations of eligibility or entry criteria.
146. Cardiorentis (and/or its contractors) conducted eleven co-monitoring visits of trial sites during the course of the TRUE-AHF trial. These were as follows.
|
Site |
Co-monitor(s) |
Dates of visit |
Date of Report |
|
0701 |
Dr Matthias Rother (IMR Consulting) |
9 July 2013 |
19 July 2013 |
|
1208 |
Dr Henri van Langenberghe |
18 September 2013 |
24 September 2013 |
|
0803 |
Elmar Schnee |
4 October 2013 |
8 October 2013 |
|
1707 |
Dr Henri van Langenberghe |
9 December 2013 |
13 January 2014 |
|
2006 |
Dr Matthias Rother (IMR Consulting) |
5 February 2014 |
7 April 2014 |
|
2003 |
Dr Matthias Rother (IMR Consulting) |
6 February 2014 |
7 April 2014 |
|
2301 |
Dr Teiba Al-Haboubi |
28 March 2014 |
4 June 2014 |
|
1806 |
Jürgen Rohner |
23 April 2014 |
4 June 2014 |
|
1803 |
Jürgen Rohner |
24 April 2014 |
26 May 2014 (draft) |
|
1205 |
Jürgen Rohner |
6 - 7 August 2014 |
9 September 2014 |
|
2505 |
Jürgen Rohner |
29-30 October 2014 |
12 November 2014 |
147. Cardiorentis (and/or its contractors) also conducted audits of the sites and IQVIA's systems during the course of the TRUE-AHF trial. These were as follows.
|
Site / System |
Auditor(s) |
Dates of audit |
Date of Report |
|
CRO System
Basel, Switzerland |
Adamas Consulting |
12 - 14 June 2013 |
15 July 2013 |
|
1503 |
Tachris AG |
24-25 July 2013 |
17 September 2013 |
|
0902 |
Tachris AG |
6 –7 August 2013 |
13 November 2013 |
|
1807 |
Tachris AG |
22-23 August 2013 |
11 October 2013 |
|
Pharmacovigilance System
Durham, North Carolina |
Adamas Consulting |
21-22 October 2013 |
15 May 2014 |
|
e-TMF
Milan, Italy |
SwissPharmAudit |
20-22 November 2013 |
10 January 2014 |
|
0424 |
Dr Jürg Lustenberger and SwissPharmAudit |
3-4 February 2014 |
8 March 2014 |
|
0434 |
Dr Jürg Lustenberger and SwissPharmAudit |
5-7 February 2014 |
8 March 2014 |
|
1605 |
Dr Jürg Lustenberger and SwissPharmAudit |
13-14 February 2014 |
31 March 2014 |
|
1802 |
SwissPharmAudit and Tachris AG |
3-5 March 2014 |
28 March 2014 |
|
2008 |
SwissPharmAudit |
6-7 March 2014 |
17 April 2014 |
|
CEVA
Durham, North Carolina |
Dr Jürg Lustenberger
|
13-14 March 2014 |
27 May 2014 |
|
0414 |
Dr Jürg Lustenberger |
2-3 April 2014 |
26 May 2014 |
|
Data Management
Illkirch |
Dr Jürg Lustenberger |
11-12 June 2014 |
24 July 2014 |
|
Quintiles Central Laboratories
Marietta, Georgia |
Dr Jürg Lustenberger |
15-16 January 2015 |
26 February 2015 |
|
1906 |
Dr Jürg Lustenberger |
20-21 January 2015 |
19 February 2015 |
|
0101 |
Dr Jürg Lustenberger |
1-2 October 2015 |
3 November 2015 |
|
0105 |
Dr Jürg Lustenberger |
5-6 October 2015 |
4 November 2015 |
|
0201 |
Dr Jürg Lustenberger |
13-14 October 2015 |
9 November 2015 |
148. IQVIA also conducted audits, for cause inspections and risk analysis audits of its systems during the TRUE-AHF trial. These were as follows.
|
Site / System |
Auditor(s) |
Dates of audit |
Date of Report |
|
0908 |
[Tracy Duggan] |
29 - 30 April 2013 |
16 May 2013 |
|
In trial audit |
Sarah Rowe |
7 - 8 May 2013 |
16 May 2013 |
|
Quality Planning team |
Veronica Sandström |
17 - 18 October 2012 |
12 November 2012 |
|
2501 |
Stella Fragou |
20 - 21 January 2014 |
4 February 2014 |
|
0422 |
Karen Whitman |
13-14 January 2015 |
28 January 2015 |
|
CAPA implementation |
Kathrin O'Hagan |
23 February 2015 - 5 May 2015 |
8 May 2015 |
149. Certain regulatory authorities conducted GCP and systems inspections during the course of the TRUE-AHF trial. In particular:
· The Netherlands Healthcare Inspectorate inspected both Cardiorentis and IQVIA during a GCP-inspection at Quintiles B.V. Hoofddorp. The inspection of IQVIA took place on 25-26 September 2013, and the inspection of Cardiorentis took place on 30 September 2013. The final inspection reports were issued by the Inspectorate on 19 December 2013.
· The German regulator in Hessen conducted a GCP inspection of site 1211 on 12 February 2014. The final inspection report was issued on 27 February 2014.
· The Medicines and Medical Devices Agency of Serbia conducted a GCP inspection of site 2006 on 4-7 November 2014. The final inspection report was issued on 13 November 2014.
· The Swiss Agency for Therapeutic Products Clinical Trials Division, GCP / PV Inspectorate ("SwissMedic") conducted a GCP inspection of site 2301 on 27-28 May 2015, and a clinical trials specific systems inspection of IQVIA on 1-2 July 2015. The final report for the inspection of site 2301 was issued on 8 July 2015 and for the systems inspection was issued on 28 July 2015.
· The FDA conducted an inspection at site 0401 between 27 June and 15 July 2016. The final report, FDA 483 Form, was issued on 15 July 2016.
· The FDA also conducted an inspection at site 0434 in April 2014 and the Chilean regulatory authority conducted an inspection at site 0301 in October 2014.
150. During the course of the TRUE-AHF trial, Cardiorentis conducted a remote data review of the information within InForm, also making use of the PD Logs and listings of SAEs. The process was described in a Cardiorentis document called a Study Specific Working Instruction dated 20 May 2015. This said, in part:
‘As part of its oversight activities, [Cardiorentis] conducts remote data review of pre-selected investigational sites and patients randomized in TRUE-AHF. … As stated in the [Cardiorentis] oversight plan for [the study], the target is to review about 20% of all patients enrolled. …
Process:
The patient data review by the Sponsor is carried out on data available remotely in read only access.
…
Quality Assessment of Individual Patient Data Review in eCRF
… The data review … is focused on consistency in completeness of all required data entries, within eCRF, trends in potentially missed endpoints, adverse events and/or protocol deviations….
In addition, planned treatment, concomitant medication and medication list at hospital discharge / day30 are checked for completeness and consistency. Within the review of ConMed section, any signs of increased AHF therapy and forbidden medication are closely reviewed. …’
DSMB Recommendations
151. On 14 March 2015 the DSMB conducted the first interim analysis. Based on this, the DSMB recommended no change in sample size and that the study should continue according to the Protocol.
152. On 16 November 2015, the DSMB conducted the second interim analysis in relation to endpoint 2, using a conditional power methodology. Based on this, the DSMB recommended to stop the trial in relation to endpoint 2 for futility, ‘as the conditional power for a positive result on cardiovascular mortality is extremely low’. Cardiorentis announced the End of Study on 19 November 2015.
The Blind Data Review
153. A matter of importance to the case was the blind data review (sometimes called the ‘BDR’) which was conducted of the study data after the conclusion of enrolment and prior to unblinding. Ultimately, and although this had not been the case at earlier stages of the litigation, there was little dispute as to the sequence of events in relation to this review or as to its results.
154. A blind data review is a process which is envisaged and provided for in the ICH-GCP. ICH-GCP E9 (Statistical Principles for Clinical Trials), contains the following guidance relevant to a blind data review process.
(a) The Glossary of ICH-GCP E9 defines the ‘Blind Review’ as:
‘The checking and assessment of data during the period of time between trial completion (the last observation on the last subject) and the breaking of the blind, for the purpose of finalising the planned analysis.’
(b) Section 5.1 of ICH-GCP E9, headed, ‘Prespecification of the Analysis’, states, inter alia, as follows:
‘When designing a clinical trial the principal features of the eventual statistical analysis of the data should be described in the statistical section of the protocol. This section should include all the principal features of the proposed confirmatory analysis of the primary variable(s) and the way in which anticipated analysis problems will be handled. In case of exploratory trials this section could describe more general principles and directions.
The statistical analysis plan … may be written as a separate document to be completed after finalising the protocol. In this document, a more technical and detailed elaboration of the principal features stated in the protocol may be included …. The plan may include detailed procedures for executing the statistical analysis of the primary and secondary variables of the data. The plan should be reviewed and possibly updated as a result of the blind review of the data … and should be finalised before breaking the blind. Formal records should be kept of when the statistical analysis plan was finalised as well as when the blind was subsequently broken…
If the blind review suggests changes to the principal features stated in the protocol, these should be documented in a protocol amendment. Otherwise, it will suffice to update the statistical analysis plan with the considerations suggested from the blind review. Only results from analyses envisaged in the protocol (including amendments) can be regarded as confirmatory.
In the statistical section of the clinical study report the statistical methodology should be clearly described including when in the clinical trial process methodology decisions were made (see ICH E3).”
(c) Section 5.2 of ICH-GCP E9 gives guidance on the consideration of analysis sets and states inter alia as follows:
‘The set of subjects whose data are to be included in the main analyses should be defined in the statistical section of the protocol. In addition, documentation for all subjects for whom trial procedures (e.g. run-in period) were initiated may be useful. The content of this subject documentation depends on detailed features of the particular trial, but at least demographic and baseline data on disease status should be collected wherever possible.
If all subjects randomised into a clinical trial satisfied all entry criteria, followed all trial procedures perfectly with no losses to follow-up, and provided complete data records, then the set of subjects to be included in the analysis would be self-evident. The design and conduct of a trial should aim to approach this ideal as closely as possible, but, in practice, it is doubtful if it can ever be fully achieved. Hence, the statistical section of the protocol should address anticipated problems prospectively in terms of how these affect the subjects and data to be analysed. The protocol should also specify procedures aimed at minimising any anticipated irregularities in study conduct that might impair a satisfactory analysis, including various types of protocol violations, withdrawals and missing values. The protocol should consider ways both to reduce the frequency of such problems, and also to handle the problems that do occur in the analysis of data. Possible amendments to the way in which the analysis will deal with protocol violations should be identified during the blind review. It is desirable to identify any important protocol violation with respect to the time when it occurred, its cause and influence on the trial result. The frequency and type of protocol violations, missing values, and other problems should be documented in the clinical study report and their potential influence on the trial results should be described (see ICH E3).
Decisions concerning the analysis set should be guided by the following principles: 1) to minimise bias, and 2) to avoid inflation of type I error.’
(d) Section 5.2.1 of ICH-GCP E9 headed ‘Full Analysis Set’ states inter alia as follows:
“The intention-to-treat … principle implies that the primary analysis should include all randomised subjects. Compliance with this principle would necessitate complete follow-up of all randomised subjects for study outcomes. In practice this ideal may be difficult to achieve, for reasons to be described. In this document the term ‘full analysis set’ is used to describe the analysis set which is as complete as possible and as close as possible to the intention-to-treat ideal of including all randomised subjects. Preservation of the initial randomisation in analysis is important in preventing bias and in providing a secure foundation for statistical tests. In many clinical trials the use of the full analysis set provides a conservative strategy. Under many circumstances it may also provide estimates of treatment effects which are more likely to mirror those observed in subsequent practice.”
(e) Section 5.2.2 of ICH-GCP E9 headed ‘Per Protocol Set’ states inter alia as follows:
‘The ‘per protocol’ set of subjects, sometimes described as the ‘valid cases’, the ‘efficacy’ sample or the ‘evaluable subjects’ sample, defines a subset of the subjects in the full analysis set who are more compliant with the protocol and is characterised by criteria such as the following:
i) the completion of a certain pre-specified minimal exposure to the treatment regimen;
ii) the availability of measurements of the primary variable(s);
iii) the absence of any major protocol violations including the violation of entry criteria.
The precise reasons for excluding subjects from the per protocol set should be fully defined and documented before breaking the blind in a manner appropriate to the circumstances of the specific trial.
The use of the per protocol set may maximise the opportunity for a new treatment to show additional efficacy in the analysis, and most closely reflects the scientific model underlying the protocol. However, the corresponding test of the hypothesis and estimate of the treatment effect may or may not be conservative depending on the trial; the bias, which may be severe, arises from the fact that adherence to the study protocol may be related to treatment and outcome.
The problems that lead to the exclusion of subjects to create the per protocol set, and other protocol violations, should be fully identified and summarised. Relevant protocol violations may include errors in treatment assignment, the use of excluded medication, poor compliance, loss to follow-up and missing data. It is good practice to assess the pattern of such problems among the treatment groups with respect to frequency and time to occurrence.’
(f) Section 5.2.3 of ICH-GCP E9 headed ‘Roles of the Different Analysis Sets’ states inter alia as follows:
‘In general, it is advantageous to demonstrate a lack of sensitivity of the principal trial results to alternative choices of the set of subjects analysed. In confirmatory trials it is usually appropriate to plan to conduct both an analysis of the full analysis set and a per protocol analysis, so that any differences between them can be the subject of explicit discussion and interpretation. In some cases, it may be desirable to plan further exploration of the sensitivity of conclusions to the choice of the set of subjects analysed. When the full analysis set and the per protocol set lead to essentially the same conclusions, confidence in the trial results is increased, bearing in mind, however, that the need to exclude a substantial proportion of subjects from the per protocol analysis throws some doubt on the overall validity of the trial.
The full analysis set and the per protocol set play different roles in superiority trials (which seek to show the investigational product to be superior), and in equivalence or non-inferiority trials (which seek to show the investigational product to be comparable, see section 3.3.2). In superiority trials the full analysis set is used in the primary analysis (apart from exceptional circumstances) because it tends to avoid over-optimistic estimates of efficacy resulting from a per protocol analysis, since the non-compliers included in the full analysis set will generally diminish the estimated treatment effect. However, in an equivalence or non-inferiority trial use of the full analysis set is generally not conservative and its role should be considered very carefully.’
155. In keeping with this guidance, in the TRUE-AHF trial it was intended that the primary analysis should be on the FAS and that there would be a secondary analysis on a PP or per protocol set that would be produced as a result of a blind data review. As already set out, each version of the Protocol section 7.1.2.1, headed ‘Study Populations’, provided that the ‘PP analysis set will be defined as patients in the FAS who do not have any major protocol deviations during the study treatment. Protocol deviations will be identified and documented following a blind data review prior to database lock.’
156. More detailed provision for the conduct of the blind data review and the production of the per protocol set was set out in the study’s Statistical Analysis Plan. As already referred to, there were two versions of the Statistical Analysis Plan which were produced before the series of blind data review meetings commenced.
157. Version 1 of the Statistical Analysis Plan, issued on 25 September 2014, had included the following, in Section 5.3, as to the Per Protocol Analysis Set:
‘The per-protocol (PP) analysis set will contain all patients in the FAS who did not experience any major protocol deviations during the study treatment. Major protocol deviations will be identified and documented following a blind data review following database lock. Protocol violations to be examined at the blind data review include:
· Failure of any inclusion/exclusion criteria
· Did not complete the 48 hours of study treatment
· >12 h between initial clinical assessment and start of study drug infusion
· Received a concomitant medication during study treatment which may affect the efficacy of study treatment
· Received study treatment other than the one to which they were randomized
Full details of the definition of major protocol violations will be included in the Blind Data Review (BDR) plan.’
158. Version 2 of the Statistical Analysis Plan, issued on 16 February 2015 did not make material changes to these provisions.
The conduct of the Blind Data Review
159. Before the Blind Data Review commenced, on 15 September 2015, Claire Raskino, who was or was shortly to become IQVIA’s Statistical Lead, raised internally the possibility that BIOS could assist in identifying protocol deviations by programmatic computer searches. She then raised this possibility at a meeting with Cardiorentis on 22 September 2015, having referred to the fact that Teiba Al-Haboubi had recently made ad hoc requests for BIOS to program listings to assist in identifying potential IC6 and EC3 protocol violations.
160. On 18 November 2015, Henri van Langenberghe emailed Jason Turner, Veronique Mahaux, Rafal Ziecina and Claire Raskino at IQVIA, with copies to Teiba Al-Haboubi, Richard Holcomb and Markus Meyer, to discuss the constitution of the per protocol population. In his email, he stated:
‘As you know, SAP Section 5.3 says that the per-protocol (PP) analysis set will contain all patients in the FAS who did not experience any major protocol deviations during the study treatment. The SAP language was intentionally made general. The goal of the PP analysis, is to get a "clean" estimate of the treatment effect, without confounding factors. We have some flexibility in defining the PP population as long as it is done in a blinded manner and not favouring one treatment arm.
And these are the violations the SAP says are to be included in the blind data review:
• Failure of any inclusion/exclusion criteria
• Did not complete the 48 hours of study treatment
• >12 h between initial clinical assessment and start of study drug infusion
• Received a concomitant medication during study treatment which may affect the efficacy of study treatment
• Received study treatment other than the one to which they were randomized
I suggest we need to start thinking about any other, if any, criteria should be included.’
The email then said that it was possible partly to match the protocol deviation categories in the PD Trend Logs with these ‘PP disqualifying deviations’, but added:
‘However, the PD log is inconsistent in the description of deviations and the level of detail given. Plus, there is no guarantee, that all PDs are captured. This means we may have to do a for purpose data extraction.’
Dr van Langenberghe ended his email by suggesting that IQVIA and Cardiorentis arrange a telephone conference to start discussions on how to proceed, preferably in the first week of December 2015.
161. In the event, the Blind Data Review ‘kick off meeting’ occurred on 9 December 2015. It was arranged by Jason Turner, and attended by him and, amongst others, Claire Raskino, Rafal Ziecina, Aline Ron and Lara Queiroz for IQVIA. The Cardiorentis attendees were or included Dr van Langenberghe and Richard Holcomb. In the slides which Claire Raskino prepared for that meeting, it was explained that BIOS had the capacity of writing programmes to identify major protocol deviations that might not have been captured in CTMS, and that BIOS-identified and CTMS-identified potential major protocol deviations might be combined into one spreadsheet, ‘often with much overlap between the two sources’, with a view to assessing which patients should be excluded from the per protocol population. The slides further stated that Claire Raskino would draft a Blind Data Review plan for the team to review.
162. On 16 December 2015 Dr van Langenberghe sent an email to Claire Raskino in which he said that he ‘took a look at the CTMS PD log … with a view to determining whether or not this log can be used for the PP population determination’, and commented that the PD log could not be used for per protocol exclusion determination regarding IP [viz Investigational Product] compliance and ‘in extension it is unlikely that it can be used for the PP population exclusion regarding any other variable to be considered.’
163. Thereafter, and in collaboration with Cardiorentis’s team as to what the listings should be, the IQVIA BIOS team wrote programmes to identify within the eCRF database a selection of potential protocol deviations which might warrant the exclusion of subjects from the per protocol population. Amongst the listings were ones for prohibited medications prior to starting Study Treatment infusion and within 72 hours after starting infusion, and non-compliance with the Protocol as to the amount of Study Treatment received.
164. One of the listings (Listing 9) about which there was a considerable amount of discussion was in relation to Inclusion Criterion 6. As a subject for a listing this was first mentioned in the draft of the Blind Data Review plan on 3 February 2016. Thereafter a number of exploratory listings for this Inclusion Criterion were produced, including a long listing of 623 pages, which was shared with the Cardiorentis team on 9 February 2016. A further draft of this listing was provided to the IQVIA and Cardiorentis BDR teams on 12 February 2016. This involved three separate parts: (1) patients who did not receive IV furosemide or equivalent diuretic at ≥40 mg at least 2 hours prior to randomisation; (2) patients who had started an IV diuretic within < 2 hours prior to randomisation; and (3) patients who had started or re-started or ended any other IV infusion of a medication (excluding diuretics) potentially to treat HF within <2 hours prior to randomisation. That listing had 600 patients who fell within one or more of those categories.
165. A face to face meeting between the two BDR teams took place on 18 and 19 February 2016 at the IQVIA office in Edinburgh. Jason Turner chaired the meeting, and Claire Raskino, Lara Queiroz, Aline Ron, Michael Abbs, Veronique Mahaux, Rafal Ziecina and Guilia Papalini were present from IQVIA. Markus Meyer, Teiba Al-Haboubi and Richard Holcomb were there from the Cardiorentis side. There was a discussion, the details of which are not clear, as to various of the Protocol Deviation listings and there was a review of the combined CTMS / BIOS Protocol Deviation spreadsheet, with specific regard to at least some of the patients on it.
166. The discussions between the teams up to that point, and at the Edinburgh meeting, were reflected in the revised draft Blind Data Review plan which Claire Raskino produced on 24 February 2016. By this stage it had been decided that BIOS PD programming would not be used to seek to identify patients who had not received IV furosemide (or equivalent diuretic) at any time after start of emergency services (or what has been called the ‘first part’ of IC6). The draft Blind Data Review plan stated that this was because it was considered that lack of a relevant eCRF entry might be a data entry failure rather than evidence that the relevant amount of IV diuretic was not administered. In addition there was a decision to start to explore the time period between randomisation and the start of study drug infusion, rather than only the two hours before randomisation referred to in IC6.
167. On 28 February 2016, Rafal Ziecina suggested to the IQVIA and Cardiorentis BDR teams that the per protocol set should include those patients with Protocol Deviations in relation to IC6 where the non-compliant dose / dose change had occurred within two hours of randomisation but not within two hours of the start of study drug infusion. This change, he said, would reduce the lists of those patients to be excluded from the PP set significantly. On 29 February 2016 Dr Ziecina emailed Ms Raskino suggesting that if this change was made the whole relevant PD listing would be reduced by 50% ‘(or even more)’ and that this would make the listing ‘very simple’. Dr Ziecina said that he hoped that Professor Meyer would agree to it. Ms Raskino suggested that Dr Ziecina should take this up with Professor Meyer which, later that day, he did, saying that his proposal seemed to be ‘the most reasonable approach’, and asking Professor Meyer whether he agreed with it.
168. At a WebEx meeting on 1 March 2016 between the IQVIA and Cardiorentis BDR team members and IQVIA BIOS programmers it was agreed that while changes in IV HF treatments within two hours of randomisation were major Eligibility PDs, it was changes in IV HF treatments within two hours of study drug infusion which would be treated as the basis upon which to exclude patients from the PP analysis set.
169. On 3 March 2016 IQVIA and Cardiorentis held the first part of a formal BDR meeting. The second part of this meeting occurred on 9 March 2016. At that meeting Claire Raskino presented the PD listings that were ready, and the combined CTMS/BIOS PD spreadsheet, with a summary which showed that at that point ‘441 randomised patients (20%) are excluded from PP population due to PDs in CTMS and/or identified by BIOS’.
170. The draft of the BDR plan produced on 9 March 2016 recorded that while PD listing 9a would be of patients who ‘started or ended any IV diuretic or other IV infusion of a medication potentially to treat HF within <2 hours prior to randomization’, these PDs were ‘not necessarily a reason to exclude from the PP’; while PD listing 9b would be for patients who ‘started or ended any IV diuretic or other IV infusion of a medication potentially to treat HF within < 2 hours prior to’ start of study drug infusion, and these would be excluded from the PP population.
171. Database lock occurred on 11 March 2016. On the following day, a new draft of the BDR Plan was produced by the IQVIA team. It included a new section which set out the rationale for excluding subjects from the PP population.
172. On 14 March 2016 a further WebEx meeting took place between the BDR teams. Key topics to be discussed were recorded in the agenda as being: re-running the PD listings using the blinded hard-locked data; updating and finalising the PD spreadsheet; and determining the numbers to be excluded from the PP population.
173. On 15 March 2016 Dr van Langenberghe sent final amendments to the draft BDR Plan. There was also another WebEx meeting between the teams. Subsequently on that day Lara Queiroz emailed the Cardiorentis team saying that their amendments had been accepted. She recorded that agreement had been reached that BIOS listings (rather than BIOS and PD logs from CTMS) would be the exclusive source for identification of all protocol deviations involving prohibited medications, including in relation to EC3. In the same email she further recorded the final decision in relation to IC6, which was that, for the furosemide requirement, only patients who were identified in the CTMS PD log as not having received furosemide of ≥ 40 mg or equivalent would be excluded, thus confirming that the BIOS listing would not be used for this part of IC6. Later again Ms Queiroz sent out what she believed would be the final PD spreadsheet.
174. On 16 March 2016, Dr Al-Haboubi raised certain questions. There was a final WebEx meeting between the BDR teams. Ms Raskino sent out a revised version of the BDR Plan and Ms Queiroz sent out a revised version of the PD spreadsheet. There were some further comments on the spreadsheet from Ms Raskino and Mr Shaw, and that led to Ms Queiroz sending out a further revised version 3 of the PD Spreadsheet. Cardiorentis representatives approved and signed the revised version of the BDR Plan, the Statistical Analysis Plan, and approved the revised Spreadsheet. After that, Cardiorentis approved the unblinding of the data.
The finalised Blind Data Review Plan
175. The BDR Plan, as revised and approved on 16 March 2016, sought to recap and summarise the BDR process which had taken place.
176. The purpose of the BDR was described at section 1 inter alia as follows:
‘Objective A: Defining and identifying major protocol violations/deviations (PV/PDs) for the purpose of establishing the analysis populations prior to database un-blinding.
Objective B: Identifying any unresolved data issues (accuracy, consistency, outlying, missing, un-coded) in the run up to database lock.
Objective C: Discuss any data issues which may affect the efficacy analysis or may require revision of the Statistical Analysis Plan (SAP) prior to database un-blinding.’
177. The Core Review Team for BDR purposes were identified at section 2. They were Claire Raskino, Rafal Ziecina, Aline Ron, Guilia Papalini, and Lara Queiroz for IQVIA; and Henri van Langenberghe, Richard Holcomb, Markus Meyer, and Teiba Al-Haboubi for Cardiorentis. Section 3 set out an overview of the process which had been followed.
178. Section 7.1 was an overview of the criteria for exclusions from the per protocol population, which were summarised in table 1. It was stated that ‘Subjects with these exclusion criteria will be identified as reported in the CTMS PD log and/or via BIOS programming to identify PD criteria in the eCRF data. Some potential exclusion criteria will require a review and decision from the BDR Team, while other criteria will automatically exclude subjects from the PP population.’ Of relevance, within table 1 it was stated that:
(a) For PD Category 2 of ‘Eligibility Criteria’, with the description ‘Non-compliance with entry criteria’, the relevant Protocol Sections were identified as 3.2.1 - 3.2.2. The proposed source was stated to be ‘CTMS (& BIOS programs for prohibited meds, missing/invalid screening BNP/NT-pro BNP and eCRF CONMED evidence that inclusion criterion 6 was not met)’. The BDR Team Review column was marked “Y” (i.e. yes). Insofar as this is read as saying that the source for eligibility deviations in patients who had received prohibited medications was CTMS and BIOS, it was not an accurate description as to how the PD Spreadsheet was produced, because of the recent agreement that only BIOS listings should be used for this purpose.
(b) For the PD Category of ‘Concomitant Medications’, with the description ‘Prohibited medication received during the first 72 hours following t0’, the Protocol Section was identified as 4.10. The proposed source was stated to be ‘BIOS’ and the BDR Team Review column was marked “N” (i.e. no).
179. Section 7.2 stated:
‘Major PDs excluding patients from the Per Protocol population will be documented in the PD Spreadsheet. The rationale for the criteria are described in Section 7.2.1. The PD Spreadsheet will be a combined version of the CTMS PD log spreadsheet and a BIOS programmed PD spreadsheet, as the two sources of identifying PDs for PP exclusions (see Table 1). For potential major PDs requiring review a column will be included in the spreadsheet to document the final BDR team decision.
In general, the ULA01 BDR team approach to the PP population will be to exclude patients only if proven ineligible, either by CRA source data verification and reporting in CTMS or by data entry in eCRF, rather than to include patients only if proven eligible. For example, lack of eCRF evidence to support an eligibility criterion (such as missing eCRF CONMED page IV diuretic entries to support inclusion criterion 6) may be a PD for data entry. However, this may not be proof that the required amount of IV diuretic was not administered at least two hours before randomization. Unless specifically stated in the CTMS PD log that this component of inclusion criterion 6 was not met, missing appropriate IV diuretic records in CONMED may not result in a confirm PDV02 failed eligibility finding and the patient may still be included in the PP population.’
180. Section 7.2.1 was headed ‘Rationale for excluding a subject from the Per Protocol Population’. Under this heading it was said, inter alia:
(1) That subjects would be excluded from the per protocol analysis if ‘there is evidence, either by verification of data reported in INFORM (via one of the listings run by Bios) or by protocol deviation reported in CTMS by the site monitor after an on-site monitoring visit (and reported as a protocol deviation in CTMS), that one or more inclusion criterion was not met or that one or more exclusion criterion were met’. Paragraph b.1) qualified this by saying that some cases were reviewed individually, and listed four categories (of which prohibited medications constituting an eligibility deviation was not one). Once again, this text can be said not perfectly to reflect what had been agreed would be, and which was, the way in which the PD Spreadsheet was compiled and the per protocol population identified, in that for such deviations only BIOS was used as the relevant source.
(2) Amongst the categories which it was said were ‘reviewed individually’ was a category of subjects whose vital signs (blood pressure, body temperature) were not measured at the time of randomisation, but were measured after randomisation, but before the start of the infusion. The BDR Plan stated that, for these patients, if the values were not within the required range, the subject was excluded from the per protocol population; if they were within the required ranges the subject was considered eligible. The rationale was that the Protocol intended to have the start of infusion drug straight after the randomization, and if there was a time lag between the randomization and the start of infusion of the study drug but nevertheless the vital signs were measured and confirmed within the required ranges, the subject was to be considered eligible.
(3) Another of the categories said to have been reviewed individually related to Creatinine clearance (Glomural Filtration Rate). It was stated that subjects with PDs reported in CTMS that stated that the subject violated the Protocol because creatinine clearance (filtration rate) was lower than 30 mL/min/1.73m² (as measured by the MDRD formula) at the time of screening were only excluded from the per protocol analysis if the actually reported value was below 25 mL/min/1.73m². This was because the relevant exclusion criterion had been updated to the latter value in Protocol amendment 3 with the intention of improving the characterization of the risk-benefit profile of ularitide in patients with more advanced renal failure, a subgroup of patients which might benefit particularly from a therapy like ularitide.
(4) A further category said to have been ‘reviewed individually’ was in relation to IC6. This recorded that only subjects who had appeared in a CTMS PD log as not receiving furosemide and/or those who appeared in a BIOS listing as having started or ended any IV diuretic or other IV infusion of a medication potentially to treat HF within < 2 hours prior to the start of infusion of the study drug were excluded from the per protocol population. The rationale for this was that the intention of IC 6 was to make sure that the subject was still symptomatic at the time of the start of the infusion of the study drug, despite having received furosemide or equivalent after the start of emergency services, and had not received, within two hours of the start of infusion of the study drug, any medication that could confound the effects of the study treatment.
(5) A further identified category of exclusion from the PP population was where there was evidence, by verification of data reported in INFORM via a BIOS listing that the subject used prohibited medication during the first 72 hours following the start of study drug infusion. The rationale for this was that use of medication prohibited by the Protocol within 72 hours of the start of the study drug might confound the effects of the study treatment.
181. Section 7.2.3 defined the major PDs identified by BIOS Programs. In relation to Listings 9a and 9b, it was stated, as it had been in drafts of the BDR Plan, that 9a would be a list of patients who had started or ended any IV diuretic or other IV infusion of a medication potentially to treat HF within < 2 hours prior to randomisation, but that this was not a reason to exclude from the PP set; while listing 9b would include patients with such changes occurring within < 2 hours of the start of infusion, which would be a reason to exclude.
182. Section 7.2.4 described the process for combining the CTMS and BIOS PD Spreadsheets.
183. The PD Spreadsheet which had been produced on 16 March 2016 was replaced by a somewhat modified one, after unblinding, on 22 March 2016. That Spreadsheet had 2311 rows of protocol deviations in respect of some 1016 different patients. The Spreadsheet also indicated whether the deviation was such as to lead to exclusion from the PP set.
The Results revealed
184. After unblinding, IQVIA prepared the TLFs (Tables Listings and Figures) giving the results of the study. The analysis of co-primary endpoint 1, for the FAS and using only observed data, was prioritised. Ms Raskino sent the result of this analysis to Cardiorentis on 22 March 2016. On 24 March 2016 IQVIA sent Cardiorentis the rest of the Headline TLFs. The non-Headline TLFs were shared with Cardiorentis on 15 April 2016.
185. The full results of the trial are referred to further below. At present it suffices to say that the TLFs contained the following:
(a) That the total number of patients enrolled and randomised and who formed the Intention to Treat set was 2157. 29 of those patients had not started study drug infusion and were therefore not in the ‘As Treated’ set.
(b) That the Primary Analysis of Co-Primary Efficacy Endpoint 1, with imputation for missing data, was as follows
|
Outcome |
Statistic |
Ularitide
N= 1088 |
Placebo
N=1069 |
Ularitide vs Placebo |
|
Improved |
n (%) |
520.9 (47.9) |
503.4 (47.1) |
|
|
No Change |
n (%) |
494.7 (45.5) |
476.9 (44.6) |
|
|
Worsened |
n (%) |
72.3 (6.6) |
88.7 (8.3) |
|
|
|
p-value |
|
|
0.830 |
(c) That a sensitivity analysis of Co-primary Efficacy Endpoint 1, using observed data only, was as follows:
|
Outcome |
Statistic |
Ularitide
N= 1088 |
Placebo
N=1069 |
Ularitide vs Placebo |
|
Improved |
n (%) |
508 (48.6) |
490 (47.5) |
|
|
No Change |
n (%) |
469 (44.8) |
456 (44.2) |
|
|
Worsened |
n (%) |
69 (6.6) |
86 (8.3) |
|
|
|
p-value |
|
|
0.327 |
|
Missing |
n |
43 |
37 |
|
(d) That the Per Protocol population, established in consequence of the BDR, was of 1762 patients, ie there had been 395 exclusions from the ITT population and 366 from the As Treated population.
(e) That the analysis of Co-primary Efficacy Endpoint 1 for the per protocol analysis set, was as follows:
|
Outcome |
Statistic |
Ularitide
N= 1088 |
Placebo
N=1069 |
Ularitide vs Placebo |
|
Improved |
n (%) |
435 (49.0) |
404 (46.2) |
|
|
No Change |
n (%) |
398 (44.8) |
403 (46.1) |
|
|
Worsened |
n (%) |
55 (6.2) |
67 (7.7) |
|
|
|
p-value |
|
|
0.137 |
186. Following the receipt of these results, there was considerable disappointment within Cardiorentis. They demonstrated no major effect, and were far from meeting the preset p-value.
187. Dr Holzmeister’s evidence was that he sensed that something had gone wrong with the trial, largely because the results appeared to show a meaningful decrease in NT pro-BNP, and that in light of that he found it difficult to accept that the results of co-primary endpoint 1 were not positive. Dr Packer and Dr Holzmeister met a contact at Merck, Dan Bloomfield, who recommended that Cardiorentis engage Janet Wittes of Statistics Collaborative Inc (‘SCI’), a statistician, to assist with a forensic analysis of the clinical trial to establish whether there were data errors.
188. Ultimately IQVIA agreed that it and Cardiorentis should jointly instruct Janet Wittes, and a tripartite contract was made on 19 May 2016. Pursuant to this, Cardiorentis was required to pay an extra Eur 2.4 million in fees to IQVIA and to agree to pay for the SCI audit, while IQVIA agreed to provide access to the study database to Ms Wittes and her team.
189. In parallel with the SCI exercise, the remaining Cardiorentis team carried out a review of the clinical trial data that was available to it in eTMF and eCRF. In October 2016 the Cardiorentis team identified what Dr Holzmeister describes in his witness statement as ‘a significant discovery’, namely that the number of patients listed in the final PD Log of 16 March 2016 had been 360 patients. In his witness statement he says that ‘[t]his was a complete surprise’ to him and that he was ‘staggered by this number’. Dr Holzmeister sent the information about ineligible patients to Ms Wittes.
190. During the course of SCI’s investigations, Ms Wittes attended a meeting of Cardiorentis with Merck on 14 December 2016 and presented certain slides as to her analyses. Dr Holzmeister, at that point, forwarded to Dr Packer and to Merck what he described as the conclusion to the SCI report. It was in these terms:
‘While the data may be reliable, SCI has concerns about Quintiles's conduct of the TRUE-AHF trial on the basis the proportion of subjects randomized in violation of eligibility criteria, and the drastically different results among the eligible and ineligible populations. According to Quintiles's original sensitivity analysis of observed data only, there was no significant difference between the subjects randomized to ularitide and the subjects randomized to placebo with regard to co-primary efficacy endpoint 1, which indicates short-term clinical outcome (nominal p = 0.33). When SCI removed all ineligible subjects from its analysis of co-primary efficacy endpoint 1, subjects randomized to ularitide had significantly better outcomes compared to subjects randomized to placebo (nominal p = 0.035). When SCI limited analysis of co-primary efficacy endpoint l to the ineligible population, subjects randomized to ularitide had significantly worse short-term clinical outcomes compared to subjects randomized to placebo (nominal p = 0.022). The radical differences between the results in the eligible and ineligible population raise concerns not only about the potential missed efficacy signal in the ITT analysis of the TRUE-AHF study, but more importantly, about potential undue harm to the ineligible patients who were randomized.’
The SCI Report
191. The final SCI report entitled ‘Review of IQVIA’s analysis of data from TRUE-AHF’ was completed on and dated 21 December 2016. It set out the Data Sources employed, which included a spreadsheet of 358 subjects who did not meet the inclusion/exclusion criteria of the study, which had been forwarded to SCI by Cardiorentis. The report then set out a number of analyses of the data from the study. On pages 25-26 the report said this:
‘In November 2016, Cardiorentis notified SCI that Quintiles had randomized 358 subjects who did not meet the protocol's eligibility criteria. Cardiorentis provided SCI a list of the subjects and the criteria that had been violated. The largest number of ineligibles were violators of exclusion criterion 3: "treatment with levosimendan, milrinone, or any other phosphodiesterase inhibitor within 7 days before randomization" and inclusion criterion 6:
Persisting dyspnea at rest despite standard background therapy for ADHF (as determined by the Investigator) which must include IV furosemide (or equivalent diuretic) at >_40 mg (or its equivalent) at any time after start of emergency services (ambulance, emergency department, or hospital). At the time of randomization, the patient must still be symptomatic. In addition, the patient should not have received an IV bolus of a diuretic for at least 2 h prior to randomization and the infusion rates of ongoing IV infusions must not have been increased or decreased for at least 2 hours prior to randomization.
Cardiorentis asked SCI to evaluate co-primary endpoint 1 in the following ways:
• removing subjects who violated exclusion criterion 3; and
• removing subjects who violated inclusion criterion 6; and
• removing subjects who violated exclusion criterion 3 or inclusion criterion 6; and,
• removing all ineligible subjects.
In addition, SCI analyzed co-primary endpoint 1 in the subset of ineligible subjects. Enrollment of ineligible subjects had a noticeable impact on the results of co-primary endpoint 1 …. For example, when the analysis included all ineligibles, 47.5% of placebo subjects and 48.6% of ularitide subjects improved (nominal p-value comparing distributions = 0.33). Excluding all ineligibles decreases the proportion of placebo subjects improving to 45.8% and increases the proportion of ularitide subjects to 49.8% (nominal p- value = 0.035). Limiting the analysis to ineligible subjects shows that the difference between the groups is significant (nominal p-value = 0.022), and in favor of placebo (56.4% of placebo and 42.7% of ularitide subjects improved).’
192. The Table setting out the results of these analyses (Table 11) was as follows:
|
Full Analysis Set |
|
|
Ularitide
(n=1088) |
Placebo
(n=1069) |
Difference in % of improved Ularitide - Placebo |
Nominal
p-value |
|
Hierarchical clinical composite
(0-48 hours following randomization) |
|
|
|
0.33 |
|
Improved |
508 (48.6%) |
490 (47.5%) |
1.1 |
|
Unchanged |
468 (44.8%) |
456 (44.2%) |
|
|
Worsened |
69 (
6
.6%) |
86
(8.3%) |
|
|
Missing |
43 |
37 |
|
|
|
Full Analysis Set - Excluding subjects who violated exclusion criterion 3* |
|
|
Ularitide
(n=1024) |
Placebo
(n=1009) |
Difference in % of improved Ularitide - Placebo |
Nominal
p-value |
|
Hierarchical clinical composite
(0-48 hours following randomization) |
|
|
|
0.21 |
|
Improved |
483 (49.1%) |
459 (47.1%) |
2.0 |
|
Unchanged |
434 (44.1%) |
436 (44.7%) |
|
|
Worsened |
67 (6.8%) |
80 (8.2%) |
|
|
Missing |
40 |
34 |
|
|
|
Full Analysis Set - Excluding subjects who violated inclusion criterion 6** |
|
|
Ularitide
(n=989) |
Placebo
(n=979) |
Difference in % of improved Ularitide - Placebo |
Nominal
p-value |
|
Hierarchical clinical composite
(0-48 hours following randomization) |
|
|
|
0.16 |
|
Improved |
465 (48.7%) |
443 (46.8%) |
1.9 |
|
Unchanged |
430 (45.1%) |
422 (44.5%) |
|
|
Worsened |
59 (6.2%) |
82 (8.7%) |
|
|
Missing |
35 |
32 |
|
|
|
|
|
|
|
|
|
Full Analysis Set - Excluding subjects who violated exclusion criterion 3* or inclusion criterion 6** |
|
|
Ularitide
(n=928) |
Placebo
(n=924) |
Difference in % of improved Ularitide - Placebo |
Nominal
p-value |
|
Hierarchical clinical composite
(0-48 hours following randomization) |
|
|
|
0.10 |
|
Improved |
441 (49.3%) |
416 (46.5%) |
2.8 |
|
Unchanged |
397
(44.3%) |
403 (45.0%) |
|
|
Worsened |
57 (6.4%) |
76 (8.5%) |
|
|
Missing |
33 |
29 |
|
|
|
Full Analysis Set - Excluding all ineligible subjects |
|
|
Ularitide
(n=903) |
Placebo
(n=896) |
Difference in % of improved Ularitide - Placebo |
Nominal
p-value |
|
Hierarchical clinical composite
(0-48 hours following randomization) |
|
|
|
0.035 |
|
Improved |
435 (49.8%) |
398 (45.8%) |
4.0 |
|
Unchanged |
384 (43.9%) |
396 (45.6%) |
|
|
Worsened |
55 (6.3%) |
75 (
8
.6%) |
|
|
Missing |
29 |
27 |
|
|
|
Ineligible subjects |
|
|
Ularitide
(n=185) |
Placebo
(n=173) |
Difference in % of improved Ularitide - Placebo |
Nominal
p-value |
|
Hierarchical clinical composite
(0-48 hours following randomization) |
|
|
|
0.022 |
|
Improved |
73 (42.7%) |
92 (56.4%) |
-13.7 |
|
Unchanged |
84 (49.1%) |
60 (36.8%) |
|
|
Worsened |
14 (8.2%) |
11 (6.7%) |
|
|
Missing |
14 |
10 |
|
|
|
All p-values are two-sided calculated using the Cochran-Mantel-Haenszel test for singly ordered data, with time from first clinical evaluation to the start of study drug infusion or randomization for subjects who did not start study treatment (≤6 hrs vs >6 hrs) and baseline systolic blood pressure (<140 mmHg vs. ≥140 mmHg) as stratification variables.
* Exclusion criterion 3: Treatment with levosimendan, milrinone, or any other phosphodiesterase inhibitor within 7 days before randomization.
** Inclusion criterion 6: "Persisting dyspnea at rest despite standard background therapy for ADHF (as determined by the Investigator) which must include IV furosemide (or equivalent diuretic) at >40 mg (or its equivalent) at any time after start of emergency services (ambulance, emergency department, or hospital). At the time of randomization, the patient must still be symptomatic. In addition, the patient should not have received an IV bolus of a diuretic for at least 2 h prior to randomization, and the infusion rates of all ongoing IV infusions of medication to treat HF must not have been increased or decreased for at least 2 h prior to randomization. |
193. The conclusion, or ‘Summary of Findings’ of the finalised SCI Report was as follows:
‘As a result of SCI’s investigation, we believe that the study produced a reliable dataset from which valid conclusions may be drawn about the co-primary endpoints. Given the findings in Section 3.1.5, SCI was not confident in the accuracy of the chemistry data; however, after review of additional selected laboratory parameters, SCI did not detect any meaningful differences based on its analyses of the eCRF CHEM and HEMA data compared to the results that Quintiles reported in the HER.
While the data may be reliable, SCI has concerns about the proportion of subjects randomized in violation of eligibility criteria, and the fact that the eligible and ineligible populations showed results on co-primary endpoint 1 that were numerically in opposite directions.
Quintiles's original sensitivity analysis of observed data only showed no significant difference between the subjects randomized to ularitide and the subjects randomized to placebo with regard to co-primary endpoint 1 (nominal p-value = 0.33). When SCI removed all ineligible subjects from the analysis of co-primary endpoint 1, subjects randomized to ularitide had significantly better outcomes compared to subjects randomized to placebo (nominal p-value = 0.035). Analysis of co-primary endpoint 1 limited to the ineligible population showed that subjects randomized to ularitide had numerically worse outcomes on co-primary endpoint 1 compared to subjects randomized to placebo (nominal p-value = 0.022). These differences between the results in the eligible and ineligible population raise concerns about the interpretation of data from the ITT population.’
194. As was pointed out on behalf of IQVIA, this conclusion was expressed in terms which differed in several respects from what Dr Holzmeister had in the previous fortnight envisaged would be the conclusion, which has been quoted above.
The NEJM Article
195. In parallel with the production of the SCI report, Dr Packer, as principal author, and others, were producing an article for publication in the NEJM containing the results of the study. This process involved a number of questions being asked by the Editors (in part relaying comments by peer reviewers), with answers being given by Dr Packer. Certain of these questions and answers have assumed some prominence in relation to the debate between the parties as to the interpretability of the TRUE-AHF study.
196. Of significance are the following:
(1) The Editors stated that:
‘… it seems clear, from review of the original trial protocol (dated April 19, 2012) and the final protocol (dated December 8, 2014) that the “acute injury hypothesis” was not contemplated at the time of the original trial design, nor was the trial originally designed to test this hypothesis… Based on the comments in Amendment 1 about the acute injury hypothesis, it seems clear that this concept of the trial was adopted at the same time as the addition of a new co-primary end point to the trial - that being cardiovascular mortality. … The revised manuscript should begin with a clear indication that the original trial hypothesis was based on the demonstration of short-term symptomatic efficacy with intermediate-term safety. It should then indicate that the demonstration of a possible long-term effect on mortality in RELAX-AHF led to the revision of the TRUE-AHF hypothesis to encompass the test of the “acute injury” concept, including the potential for a reduction in acute injury to lead to a long-term survival benefit.’
In answer to these points, Dr Packer wrote (inter alia) that:
‘The revised version of the manuscript now makes clear the trial’s original hypothesis (i.e. evaluation of the short-term effects of ularitide) and the conversion of the trial shortly after its onset to a cardiovascular mortality trial, with the specific intent of testing the “acute distension-myocardial injury” hypothesis.’
(2) The Editors asked why, under the original hypothesis, ularitide had been chosen. Specific questions included: if a vasodilator was thought likely to be effective, why not test IV nitroglycerin? Was a diuretic effect anticipated, and if so why as no such effect had previously been documented in major trials of natriuretic peptides? Why choose this drug to achieve symptom relief, and, generally, ‘why should ularitide be superior to standard therapy (aggressive use of IV diuretics, perhaps most importantly) in this setting?’
To this, Dr Packer replied:
‘We did not anticipate nor have we claimed that ularitide has unique properties. Ularitide does differ from other natriuretic peptides (e.g. its endogenous source is the kidney rather than the heart, and its degradation is resistant to neprilysin). However, the selection of ularitide was not based on the uniqueness of its chemistry or physiology, but on the uniqueness of having a Sponsor with the willingness and resources to launch a large-scale trial to evaluate the drug’s efficacy. The investigators grasped the availability of this funding source as a wonderful opportunity to test an important hypothesis about acute heart failure…’
(3) The Editors commented that they believed ‘that a trial originally focused on the acute injury hypothesis would, or at least probably would, be designed differently from TRUE-AHF….’ To this Dr Packer replied:
‘Although we can understand why the editors might think that a trial originally focused on the acute injury hypothesis might be designed differently from TRUE-AHF, this is truly not the case. Our original intent (even when the trial was focused only on short-term outcomes) was to administer the study drug as early as possible, based on our belief that time was of the essence even if the endpoints were entirely short-term…’
(4) One Reviewer had commented that the conclusion in the draft paper then under consideration was misleading as it did not address the fact that two co-primary endpoints were not met. Dr Packer answered:
‘The paper has been revised to make clear that neither of the two co-primary endpoints were met.’
197. Following the completion of this process, the results of the TRUE-AHF study were ultimately published in the NEJM on 12 April 2017. Dr Packer was the lead and first-named author. 20 authors were given including Ms Wittes and, as last-named, Dr Holzmeister.
198. The Abstract of the article was as follows:
‘BACKGROUND
In patients with acute heart failure, early intervention with an intravenous vasodilator has been proposed as a therapeutic goal to reduce cardiac-wall stress and, potentially, myocardial injury, thereby favorably affecting patients’ long-term prognosis.
METHODS
In this double-blind trial, we randomly assigned 2157 patients with acute heart failure to receive a continuous intravenous infusion of either ularitide at a dose of 15 ng per kilogram of body weight per minute or matching placebo for 48 hours, in addition to accepted therapy. Treatment was initiated a median of 6 hours after the initial clinical evaluation. The coprimary outcomes were death from cardiovascular causes during a median follow-up of 15 months and a hierarchical composite end point that evaluated the initial 48-hour clinical course.
RESULTS
Death from cardiovascular causes occurred in 236 patients in the ularitide group and 225 patients in the placebo group (21.7% vs. 21.0%; hazard ratio, 1.03; 96% confidence interval, 0.85 to 1.25; P = 0.75). In the intention-to-treat analysis, there was no significant between-group difference with respect to the hierarchical composite outcome. The ularitide group had greater reductions in systolic blood pressure and in levels of N-terminal pro–brain natriuretic peptide than the placebo group. However, changes in cardiac troponin T levels during the infusion did not differ between the two groups in the 55% of patients with paired data.
CONCLUSIONS
In patients with acute heart failure, ularitide exerted favorable physiological effects (without affecting cardiac troponin levels), but short-term treatment did not affect a clinical composite end point or reduce long-term cardiovascular mortality.’
199. On the second page of the article appeared the following:
‘The first author, who had unrestricted access to the data, prepared the drafts of the manuscript, which were then reviewed and edited by all the authors, independent of the Sponsor. The authors assume responsibility for the accuracy and completeness of the analyses; the last author attests to the fidelity of the trial to the protocol.’
200. On the fourth to sixth pages of the article the following was set out:
‘The coprimary outcome of death from cardiovascular causes occurred in 236 patients in the ularitide group and 225 patients in the placebo group (21.7% vs. 21.0%; hazard ratio, 1.03; 96% confidence interval [CI], 0.85 to 1.25; P = 0.75) (Table 2 and Fig. 2). The lack of a significant difference between the two groups was seen consistently across prespecified subgroups (except for a nominally significant interaction for geographical region), as well as in subgroups that were defined according to baseline levels of NT-proBNP and cardiac troponin (Fig. S3 in the Supplementary Appendix). The distribution of responses for the clinical composite (the second coprimary outcome) did not differ significantly between the groups (Table 2, and Table S3 in the Supplementary Appendix). In a post hoc analysis that excluded the patients who had been identified before the database lock as having been ineligible for the trial, a benefit of ularitide was shown with respect to the hierarchical clinical composite outcome (P = 0.03) but not with respect to cardiovascular mortality (Table S4 in the Supplementary Appendix).
Because the tests for the two coprimary outcomes were not significant, and given the hierarchical testing plan, all secondary end-point analyses were exploratory. There was no benefit of ularitide for any of the clinical secondary outcome measures (Table 2).’
201. Table 1 to the article set out the demographic and clinical characteristics of the patients at baseline. It was as follows:
|
1. Table 1. Demographic and Clinical Characteristics of the Patients at Baseline.* |
|
Characteristic |
Ularitide (N = 1088) |
Placebo (N = 1069) |
|
Age — yr |
68.7±11.4 |
68.3±11.3 |
|
Group — no. (%) |
|
<65 yr |
362 (33.3) |
387 (36.2) |
|
≥65 yr |
726 (66.7) |
682 (63.8) |
|
Male sex — no. (%) |
714 (65.6) |
706 (66.0) |
|
Nonblack race — no. (%)† |
989 (90.9) |
973 (91.0) |
|
Body-mass index‡ |
29.3±6.3 |
29.2±6.7 |
|
Left ventricular ejection fraction — no./total no. (%) |
|
<40% |
445/690 (64.5) |
449/681 (65.9) |
|
≥40% |
245/690 (35.5) |
232/681 (34.1) |
|
Region — no. (%) |
|
North America |
159 (14.6) |
152 (14.2) |
|
Latin America |
171 (15.7) |
160 (15.0) |
|
Western Europe |
212 (19.5) |
208 (19.5) |
|
Eastern Europe |
546 (50.2) |
549 (51.4) |
|
Interval between clinical evaluation and initiation of treatment
— no. (%) |
|
≤6 hr |
533 (49.0) |
528 (49.4) |
|
>6 hr |
555 (51.0) |
541 (50.6) |
|
Clinical history — no. (%) |
|
Coronary artery disease |
556 (51.1) |
549 (51.4) |
|
Diabetes |
414 (38.1) |
429 (40.1) |
|
Previous episode of heart failure |
825 (75.8) |
806 (75.4) |
|
NYHA class within past month — no./total no. (%) |
|
I |
40/816 (4.9) |
46/830 (5.5) |
|
II |
265/816 (32.5) |
269/830 (32.4) |
|
III |
396/816 (48.5) |
398/830 (48.0) |
|
IV |
115/816 (14.1) |
117/830 (14.1) |
|
Blood pressure — mm Hg |
|
Systolic |
134.2±17.8 |
135.1±17.9 |
|
Diastolic |
79.0±13.1 |
79.4±13.5 |
|
Heart rate — beats/min |
85.4±18.8 |
85.6±19.1 |
|
Laboratory values |
|
Median N-terminal proBNP (IQR) — pg/ml |
7156
(4230–13,238) |
7121
(3974–12,599) |
|
Median cardiac troponin T (IQR) — pg/ml |
34 (22–54) |
33 (21–54) |
|
Hemoglobin — g/dl |
13.1±1.78 |
13.2±1.89 |
|
Serum creatinine — mg/dl |
1.24±0.37 |
1.23±0.35 |
|
Treatment at randomization — no. (%) |
|
Intravenous nitrates |
101 (9.3) |
110 (10.3) |
|
Intravenous dobutamine |
4 (0.4) |
6 (0.6) |
202. Table 2 summarised the results as to primary and secondary outcome measures and biomarkers as to drug response. It was as follows:
|
Table 2. Primary and Secondary Outcome Measures and Biomarkers of Drug Response. |
|
Measure |
Ularitide (N = 1088) |
Placebo (N = 1069) |
P Value |
|
Primary outcomes |
|
Cardiovascular death |
236 (21.7) |
225 (21.0) |
0.75 |
|
Hierarchical clinical composite outcome ≤48 hr after randomization — no./total no. (%) |
|
|
0.82 |
|
Improved |
508/1045 (48.6) |
490/1032 (47.5) |
|
|
Unchanged |
468/1045 (44.8) |
456/1032 (44.2) |
|
|
Worse |
69/1045 (6.6) |
86/1032 (8.3) |
|
|
Secondary outcomes* |
|
Median length of hospital stay during first 30 days (IQR) — hr |
160.8
(96.0 to 228.9) |
148.2
(94.0 to 216.8) |
0.16 |
|
Median length of stay in intensive care unit during first 120 hr (IQR) — hr |
68.0
(49.3 to 93.6) |
69.8
(50.3 to 94.3) |
0.24 |
|
No. of episodes of in-hospital worsening of heart failure during first 120 hr |
115 |
126 |
0.63 |
|
In-hospital worsening of heart failure during first 120 hr — no. (%) |
90 (8.3) |
94 (8.8) |
0.70 |
|
Rehospitalization for heart failure ≤30 days after index hospital discharge — no./total no. (%) |
75/1055 (7.1) |
74/1053 (7.0) |
1.00 |
|
Median duration of intravenous therapy for heart failure during index admission (IQR) — hr |
70.5
(42.7 to 115.4) |
68.9
(44.6 to 115.5) |
0.53 |
|
Mean (±SD) change in serum creatinine from baseline to 72 hr — mg/dl |
0.15±0.38 |
0.09±0.33 |
0.002 |
|
All-cause mortality or hospitalization for a cardiovascular cause at 6 mo — no. (%) |
443 (40.7) |
398 (37.2) |
0.10 |
|
Biomarkers of drug response |
|
Decrease in N-terminal proBNP from baseline to 48 hr |
|
No. of patients evaluated |
967 |
956 |
|
|
Median (IQR) — pg/ml |
–3816
(–7166 to –1614) |
–2595
(–5611 to –574) |
<0.001 |
|
Ratio of cardiac troponin T at 48 hr vs. baseline |
|
No. of patients evaluated |
603 |
579 |
|
|
Median (IQR) |
1.01
(0.86 to 1.19) |
1.00
(0.88 to 1.15) |
0.70 |
* The analyses of secondary outcome measures are exploratory, because the protocol specified the performance of hierarchical testing of the secondary outcomes only if the result for at least one of the coprimary outcomes was significant.
203. In the discussion section of the paper appeared the following commentary:
‘In the TRUE-AHF trial, ularitide exerted its expected short-term hemodynamic effects. The drug produced systemic vasodilation (as evidenced by decreases in systolic blood pressure), which was accompanied by decreases in NT-proBNP levels (reflecting a reduction in cardiac-wall stress). Both the hematocrit and serum creatinine levels increased during the infusion, pointing to hemo-concentration and (together with a decrease in liver enzymes indicative of less hepatic congestion) to aggressive decongestion; these effects were paralleled by a decrease in the rate of in-hospital heart-failure events during the infusion. Such early worsening events have been linked to increases in both cardiac filling pressures and cardiac troponin levels, which suggests that these events may reflect undertreated ventricular distention and acute cardiac injury at the time of initial admission. A reduction in cardiac-wall stress that is achieved rapidly after clinical presentation might be expected to reduce myocardial necrosis, preserve ventricular function, maintain clinical stability, and reduce the long-term risk of cardiovascular death. However, even though the time from clinical evaluation to pharmacologic intervention was shorter than in previous studies, and despite evidence of meaningful cardiac decongestion, the long-term risk of cardiovascular death was not reduced among patients who received ularitide. This lack of benefit raises doubt about the theories that early ventricular distention causes myocardial necrosis and adversely affect the natural history of heart failure after hospitalization and that rapid reversal of short-term ventricular distention preserves myocardial viability. For all the patients who underwent randomization, a clinical composite end point was also not affected by treatment.’
204. The authors of the NEJM article also published a Supplementary Appendix to it to provide additional information about their work. One of the pieces of further information presented was in Table S4. That Table, and accompanying text, were in these terms:
|
|
Placebo
(n=896) |
Ularitide
(n=903) |
P Value |
|
|
|
|
|
|
Cardiovascular death |
186 (20.8%) |
187 (20.7%) |
0.87a |
|
|
|
|
|
|
Hierarchical clinical composite (0-48 hours after randomization) |
0.035b |
|
Improved |
398 |
435 |
|
Unchanged |
396 |
384 |
|
Worsened |
75 |
55 |
|
Missing |
27 |
29 |
Patients who had been identified before the database lock as having been ineligible for the trial (but had still been randomized) were excluded from the above analyses.
For the hierarchical clinical composite, the favorable effect of ularitide in eligible patients (nominal P=0.035) [shown above] was directionally opposite to the unfavorable effect of the drug in ineligible patients (nominal P=0.022). These post hoc findings raise important concerns about the interpretability of primary analysis for the hierarchical clinical composite.
All p-values are 2-sided. Symbols indicate statistical methods, which are identical to Table 2 in the main paper: (a) Cox proportional hazards regression model with age, gender, baseline systolic blood pressure), time from the first clinical evaluation to start of study drug infusion or to randomization for subjects who did not start study treatment and region included as covariates, (b) Cochran-Mantel- Haenszel test for singly ordered data, with time from first clinical evaluation to start of study drug infusion or randomization for subjects who did not start treatment and baseline systolic blood pressure as stratification variables for each multiply-imputed dataset.’
205. As Supplementary Figure S1, the authors included the following diagram and accompanying text:
Supplementary Figure S1
‘The hypothesis being tested in the TRUE-AHF trial was whether intravascular congestion and ventricular distension (the events on the left depicted in green) were causally related to myocardial injury and an accelerated risk of hospitalization and death (the events on the right depicted in pink).’
The NEJM Editorial
206. Professor Paul J Hauptman wrote an Editorial in the NEJM entitled ‘Disease Modification in Acute Decompensated Heart Failure’, and which was published in the journal in May 2017.
207. In that editorial, having introduced the Packer et al paper on the results of the TRUE-AHF trial, Professor Hauptman said this:
‘The justification for the use of this particular medication is not robust, since ularitide had been evaluated in only two previous studies involving a total of 245 patients who were treated for 24 hours. In addition, although
a related natriuretic peptide, recombinant BNP (nesiritide), has been shown to have favorable short-term effects on hemodynamics and dyspnea relief, subsequent studies reported an increased risk of renal failure and a lack of survival benefit. Furthermore, hypotension was enough of a concern that patients who were enrolled in TRUE-AHF were required to have a minimum systolic blood pressure of 116 mm Hg, a criterion that unavoidably excluded the most compromised cohort of patients.’
208. Later in the Editorial Professor Hauptman said:
‘In the final analysis of the TRUE-AHF trial, no differences in the coprimary end points were observed between ularitide and placebo. The overall proportion of patients in the placebo group who had short-term worsening of their condition was quite small, which suggests that the prospect of an ultimate success for ularitide was statistically limited. However, that is not to say that ularitide had a completely neutral effect; there were fewer in-hospital heart-failure events and greater reductions in NT-proBNP levels in the ularitide group than in the placebo group.
Where does this leave us? We can conclude that ularitide, like its predecessor nesiritide, has limited short-term effects that wane after the
discontinuation of treatment, which lessens the likelihood that there is a constructive avenue for further development of natriuretic peptides. It
also appears that we do not have a mandate to establish rapid-response teams for patients who present with acute decompensated heart failure.
At this point, we should remind ourselves that the primary immediate objective of treatment is the patient-centric goal of symptom relief. In
addition, the idea that it is the hospitalization itself that increases the risk of cardiovascular death has been partially debunked, and not for the
first time. This conclusion will allow us to change the focus from disease modification so that mortality is once again relegated to a safety,
not an efficacy, end point. We also need greater consensus on how to define the response to an intervention and to determine which patients are
in greatest therapeutic need (e.g., those with in-hospital worsening despite conventional therapy).
In reality, there should be no surprise here. Exacerbations of chronic disease reflect the chronic disease, not the hospitalizations used to manage those exacerbations…’
209. A recombinant BNP, nesiritide, was initially approved in the USA in 2001 based on its acute ability to reduce pulmonary capillary wedge pressure and improve symptomatic dyspnoea. Subsequently, Johnson & Johnson conducted a trial called ASCEND-HF to examine the effects of nesiritide on patients with ADHF. The study enrolled 7141 patients with ADHF. Randomisation was completed in August 2010.
210. The results of the ASCEND-HF trial were reported in the NEJM in July 2011 (O’Connor et al). The ‘Methods’ of the trial were summarised as:
‘We randomly assigned 7141 patients who were hospitalized with acute heart failure to receive either nesiritide or placebo for 4 to 168 hours in addition to standard care. Coprimary endpoints were the change in dyspnea at 6 and 24 hours, as measured on a 7-point Likert scale, and the composite end point of rehospitalization for heart failure or death within 30 days.’
211. The ‘Results’ were summarised as follows:
‘Patients randomly assigned to nesiritide, as compared with those assigned to placebo, more frequently reported markedly or moderately improved dyspnea at 6 hours (44.5% vs. 42.1%, P = 0.03) and 24 hours (68.2% vs. 66.1%, P = 0.007), but the prespecified level for significance (P≤0.005 for both assessments or P≤0.0025 for either) was not met.
The rate of rehospitalization for heart failure or death from any cause within 30 days was 9.4% in the nesiritide group versus 10.1% in the placebo group (absolute difference, −0.7 percentage points; 95% confidence interval [CI], −2.1 to 0.7; P = 0.31).
There were no significant differences in rates of death from any cause at 30 days (3.6% with nesiritide vs. 4.0% with placebo; absolute difference, −0.4 percentage points; 95% CI, −1.3 to 0.5) or rates of worsening renal function, defined by more than a 25% decrease in the estimated glomerular filtration rate (31.4% vs. 29.5%; odds ratio, 1.09; 95% CI, 0.98 to 1.21; P = 0.11).’
212. The authors summarised their conclusions as follows:
‘Nesiritide was not associated with an increase or a decrease in the rate of death and rehospitalization and had a small, nonsignificant effect on dyspnea when used in combination with other therapies. It was not associated with a worsening of renal function, but it was associated with an increase in rates of hypotension. On the basis of these results, nesiritide cannot be recommended for routine use in the broad population of patients with acute heart failure.’
213. Based on those trial results, Johnson & Johnson stopped manufacturing nesiritide in many markets including the USA, Canada and the EU (Sager, first report para. 81(a); Joint Memorandum of Cardiology Experts, para. 11).
214. Another drug which has been studied is Novartis’s serelaxin, a recombinant form of human relaxin 2, an endogenous vasodilator involved in physiologic adaptations during pregnancy. It has similar physiologic effects to ularitide.
215. A study in relation to serelaxin, called RELAX-1, was reported in the Journal of the American College of Cardiology in January 2013 (Metra et al). As already mentioned above, that study appeared to show a reduction in chronic heart failure symptoms and a reduction in 180 day mortality. When those results were reviewed by the FDA Advisory Committee there were concerns that the results might not be real and the Committee did not support FDA approval for that indication.
216. Novartis conducted a second study, RELAX-2, between 2013 and 2017 with randomisation being completed in February of the latter year. The results were reported in the NEJM in August 2019 (Metra et al). The ‘Methods’ of this trial were summarised in this way:
‘In this multicenter, double-blind, placebo-controlled, event-driven trial, we enrolled patients who were hospitalized for acute heart failure and had dyspnea, vascular congestion on chest radiography, increased plasma concentrations of natriuretic peptides, mild-to-moderate renal insufficiency, and a systolic blood pressure of at least 125 mm Hg, and we randomly assigned them within 16 hours after presentation to receive either a
48-hour intravenous infusion of serelaxin (30 μg per kilogram of body weight per day) or placebo, in addition to standard care. The two primary end points were death from cardiovascular causes at 180 days and worsening heart failure at 5 days.’
217. The ‘Results’ were summarised as follows:
‘A total of 6545 patients were included in the intention-to-treat analysis. At day 180, death from cardiovascular causes had occurred in 285 of the 3274 patients (8.7%) in the serelaxin group and in 290 of the 3271 patients (8.9%) in the placebo group (hazard ratio, 0.98; 95% confidence interval [CI], 0.83 to 1.15; P = 0.77). At day 5, worsening heart failure had occurred in 227 patients (6.9%) in the serelaxin group and in 252 (7.7%) in the placebo group (hazard ratio, 0.89; 95% CI, 0.75 to 1.07; P = 0.19). There were no significant differences between the groups in the incidence of death from any
cause at 180 days, the incidence of death from cardiovascular causes or rehospitalization for heart failure or renal failure at 180 days, or the length of the index hospital stay. The incidence of adverse events was similar in the two groups.’
218. The authors’ conclusions were summarised thus:
‘In this trial involving patients who were hospitalized for acute heart failure, an infusion of serelaxin did not result in a lower incidence of death from cardiovascular causes at 180 days or worsening heart failure at 5 days than placebo.’
219. A further trial which requires notice is the so-called GALACTIC trial. In this trial, for which randomisation was completed in February 2018, 788 patients with acute HF and a median blood pressure of 130/75 mmHg were randomised to receive intensive, multimodal, up-titrated vasodilator therapy with nitrates, other approved vasodilators and, later in the trial, sacubitril-valsartan, versus standard care. The objective was to evaluate the effect of a strategy that emphasised early intensive and sustained vasodilation.
220. The results were published in the Journal of the American Medical Association in December 2019 (Kozhuharov et al). The primary endpoint of a composite of all-cause mortality or rehospitalization for acute heart failure at 180 days was not significantly different between the two randomised groups.
221. The authors summarised the ‘Conclusions and Relevance’ of the GALACTIC study as follows:
‘Among patients with AHF, a strategy of early intensive and sustained vasodilation, compared with usual care, did not significantly improve a composite outcome of all-cause mortality and AHF rehospitalization at 180 days.’
222. On 23 March 2018 Cardiorentis filed suit against IQVIA Ltd and IQVIA RDS Inc in the North Carolina Business Court. The claim made was that IQVIA had failed to perform its obligations in respect of the TRUE-AHF trial, ‘and then concealed its failures until it was too late to remedy them’, with the result that ‘hundreds of ineligible patients were improperly included in the Clinical Trial, fundamentally undermining the validity of the Clinical Trial and rendering it worthless’. The allegation was made that IQVIA’s failure to conduct the trial in accordance with its contractual and ethical obligations ‘was not merely the result of negligence and lack of skill, but … was the result of an intentional corporate decision to starve the Clinical Trial of resources in order to prop up Quintiles’ stock price’.
223. One feature of the North Carolina Complaint was that it alleged that IQVIA had, in the PD Logs, ‘affirmatively misrepresented’ to Cardiorentis the number of ineligible patients included in the study, that ‘it was not until the day that the data was unblinded that Quintiles disclosed to Cardiorentis that a very significant percentage of the patients enrolled in this multi-year Clinical Trial were actually ineligible to participate’, and that ‘Quintiles’ repeated and ongoing failure to identify the ineligible patients to Cardiorentis was intentional, and designed to conceal Quintiles’ poor performance and to prevent Cardiorentis from learning the true facts.’ The Complaint included a claim for punitive damages.
224. On 31 December 2018 the North Carolina Business Court stayed the North Carolina proceedings on forum non conveniens grounds.
225. On the same date, IQVIA Ltd commenced proceedings in this court against Cardiorentis (in Action CL-2018-000841) claiming in respect of what were said to be unpaid invoices relating to the TRUE-AHF trial. The sum claimed was Eur 9,501,587.24, together with interest.
Cardiorentis’s English proceedings
226. On 1 February 2019 Cardiorentis commenced proceedings in this court (Action CL-2019-000064) against IQVIA Ltd and IQVIA RDS Inc, bringing here its claims in respect of the allegedly defective performance by IQVIA of its obligations in respect of the TRUE-AHF Trial. The claim set out in the Claim Form embraced four elements: (1) breach by IQVIA of the GSA and/or of the CQA; (2) negligent breach of duty by IQVIA in the provision of services and/or in the making of representations about such services; (3) a claim under the North Carolina Unfair and Deceptive Practices Act, NCGS §75-1; and (4) a claim for injunctive relief that IQVIA should give Cardiorentis access to the data and information generated in the trial.
227. By order of Popplewell J dated 5 August 2019, the two actions, CL-2018-000841 and CL-000064, were consolidated. Cardiorentis’s claim in the action it had commenced was treated as the Part 7 claim in the consolidated action, and the claim made by IQVIA in the action which it had commenced was to be treated as a Part 20 counterclaim in the consolidated action. Cardiorentis was to be treated as the Claimant in the consolidated action, and the IQVIA entities as the Defendants.
The Issues on the pleadings
228. Cardiorentis’s claim, as pleaded in more detail in its original POC served in July 2019, included pleas to the effect that, prior to the BDR meeting on 18 and 19 February 2016, Cardiorentis had only received information in relation to eligibility deviations from the PD Logs, and that prior to 16 March 2016 Cardiorentis had not been made aware of the number of eligibility deviations identified by BIOS searches and which were included in the PD Spreadsheet.
229. As a result of IQVIA’s Defence, which had pleaded that information had been supplied during the course of the BDR process from BIOS searches, this part of Cardiorentis’s case was amended in its APOC served in February 2020.
230. Cardiorentis’s case, as set out in its APOC can be summarised as follows. Its primary pleaded claim against IQVIA was that:
(1) IQVIA was required to provide the services which it contracted to provide under the GSA with the standard of care customary in the contract research organization industry (under the express term at clause 10(a) of the GSA) and/or with reasonable care and skill (under an implied term pursuant to section 13 of the Supply of Goods and Services Act 1982); and [APOC 8(d) and 10] and
(2) in breach of these express and/or implied contractual terms, IQVIA failed to perform the services under the GSA with the required standard of care. [APOC 33]
231. The particulars of breach pleaded by Cardiorentis in respect of this claim are that IQVIA: [APOC 33]
(1) failed to train investigators adequately or at all so as to ensure that the trial was conducted in accordance with the protocol, particularly with respect to compliance with eligibility and entry criteria;
(2) failed to monitor the sites and identify protocol deviations promptly, adequately or at all;
(3) failed to conduct any or any adequate further on-site training in order to remedy existing protocol deviations and minimise the risk of further such deviations in the future;
(4) failed to take or to recommend any or any adequate other step to rectify or prevent protocol deviations;
(5) failed, in planning the study, training investigators, and monitoring sites to take any or any adequate account of the higher risk of protocol departures posed by sites in Eastern Europe (and, in particular, in countries which formed the former Soviet Union, the eastern bloc and the former Yugoslavia), of which they were or ought to have been aware;
(6) failed to provide any or any adequate training or monitoring by IQVIA medical advisors;
(7) failed to report protocol deviations to Cardiorentis in an appropriate, clear or timely manner (in particular, instances of breach of the eligibility and entry criteria);
(8) in the case of site 2006, used bulk entries for protocol deviations in such a way that the summary in the PD Trend Logs was misleading as to the rate of protocol deviations and understated the true incidence of protocol deviations concerning entry and eligibility criteria;
(9) failed to review protocol deviations to identify, escalate and remediate trends adequately or at all; and
(10) failed to ensure that sufficiently accurate information as to the nature and the reliability of the data generated by TRUE-AHF (in particular, the incidence of recruitment of ineligible subjects, other protocol deviations, and ensuring that all missing data was identified and added) was made available to Cardiorentis or that sufficient attention was drawn to the significance of the data at the Blind Data Review Meeting (or otherwise prior to unblinding of the data).
232. In support of its allegations of breach, Cardiorentis alleged that a number of ‘irregularities’ occurred across a number of sites, and in particular that ineligible patients were wrongly randomised and included in the trial, that IQVIA failed promptly, clearly or fully to identify and/or report on these irregularities to Cardiorentis and that many of them remained unreported to Cardiorentis when the Blind Data Review Meeting took place in February 2016. [APOC 20-21]
233. In particular, Cardiorentis pointed in its pleading to seven sites (2006, 2003, 0903, 1906, 0434, 1205 and 1605) in six countries (Serbia, Czechia, Romania, USA, Germany and Italy) in relation to which it was pleaded that there had been delays / inadequacies in reporting eligibility deviations.
234. In response to this part of Cardiorentis’s claim, IQVIA accepted in its Defence that the GSA included an implied term pursuant to Section 13 of the Supply of Goods and Services Act 1982, whilst contending that this did not create any obligations beyond those expressly created by clause 10(a) of the GSA because the standard of care that was reasonable for the purposes of the implied term was the standard which the parties expressly agreed pursuant to clause 10(a) of the GSA. [ADCC 10]
235. IQVIA denied that it had failed to meet the required standard of care in its performance of the services under the GSA. [ADCC 48]
236. Cardiorentis’s second pleaded claim against IQVIA was that:
(1) on a true construction of the GSA, IQVIA was required to conduct 100% SDV of the TRUE-AHF trial data, and that this entailed the verification of every data point (and of the accuracy of all data, particularly in relation to subject eligibility criteria); [APOC 9] and
(2) in breach of this obligation, IQVIA failed to conduct 100% SDV in a timely or appropriate manner (or at all) and, in any event, by the time of the BDR Meeting on 18 and 19 February 2016. [APOC 32]
237. In its Defence, IQVIA: [ADCC 9, 47]
(1) denied that it was a contractual requirement under the GSA that it carry out 100% SDV;
(2) denied that the process of SDV involves verifying the accuracy of data points, asserting that source data verification instead involves verifying only that data in the eCRF corresponded with data recorded in source documents;
(3) alleged that in any event it did carry out 100% SDV; and
(4) contended that even if there was an obligation to carry out 100% SDV, this did not amount to a guarantee by IQVIA that the SDV process would be entirely free from error.
238. Cardiorentis’s third pleaded claim against IQVIA was in respect of the CQA.
239. As part of its pleaded case on this issue Cardiorentis alleged that in circumstances where there was no express choice of law under the CQA, then pursuant to Article 4(1)(b) of Regulation (EC) 593/2008 the CQA is governed by the law of North Carolina. Alternatively, and contrary to this primary case, the CQA is governed by English law. [APOC 12]
240. Cardiorentis further alleged that:
(1) the CQA expressly provides (in clause 4.4.1) that project quality issues associated with TRUE_AHF would be processed by IQVIA’s staff in accordance with its Standard Operating Practice CS_OP_QA002: Managing Quality Issues and (in clause 4.4.2) that IQVIA was to notify critical quality issues to Cardiorentis within one business day of such issues being confirmed as critical and major quality issues within seven business days of IQVIA being notified; [APOC 13(a)-(b)] and
(2) the CQA contained an express obligation for the parties to hold monthly QA to QA meetings to deal with quality issues, audits and audit corrective and preventive actions; and [APOC 13(c)]
(3) the CQA contained an implied obligation under North Carolina law that there would be good faith and fair dealing, and that this required that neither party would do anything which injures the right of the other to receive the benefits of the agreement. Alternatively, and contrary to this primary case, if the CQA was governed by English law, it was an implied term under section 13 of the Supply of Goods and Services Act 1982 that IQVIA RDS, Inc. would exercise reasonable care and skill in carrying out the services to be provided under the CQA [APOC 14].
241. Cardiorentis then pleaded that these provisions were breached by IQVIA in the following terms:
(1) IQVIA was in breach of clause 4.4.1 of the CQA in that project quality issues were not processed in accordance with the operating procedures specified [APOC 35];
(2) IQVIA was in breach of clause 4.4.2 of the CQA in that project quality issues were not notified to Cardiorentis timeously, or even within a reasonable period and, in any event, in a statistically significant number of cases, they were not notified to Cardiorentis before the Blind Data Review Meeting [APOC 36];
(3) IQVIA was in breach of clause 5.2 of the CQA in that the QA to QA meetings which were arranged were not on a monthly basis and did not deal adequately with quality issues, audits, or audit CAPAs and project level questions or concerns [APOC 37];
(4) IQVIA was in breach of the implied term of the CQA in that they presented data in the PD Trend Logs in a way that disguised and unfairly and misleadingly understated the true incidence of protocol deviations concerning entry and eligibility criteria in a way that Cardiorentis’ right to receive the benefits of the CQA were injured. Alternatively, if English law applies to the CQA, then IQVIA failed to exercise reasonable care and skill in presenting the data in a way that understated the true incidence of protocol deviations. [APOC 38]
242. In its Defence, IQVIA denied that the CQA is governed by North Carolina law, contending instead that it is governed by English law pursuant to Article 3(1) of Regulation (EC) 593/2008, alternatively pursuant to Article 4(4) of Regulation (EC) 593/2008. [ADCC 12-15]
243. IQVIA further denied that, on its primary case that the CQA is governed by English law, it included the implied term alleged by Cardiorentis, on the basis that the CQA is not a contract under which a person agreed to carry out a service within the meaning of Section 12(1) of the Supply of Goods and Services Act 1982 [ADCC 18(3)].
244. Cardiorentis pleaded that, by reason of the breaches of contract it alleged that IQVIA had committed (in respect of the implied standard of care, the alleged express obligation to undertake 100% source data verification and the various express and implied obligations under the CQA):
(1) By the time of the BDR Meeting, Cardiorentis did not have a reliable understanding of the incidence of randomisation of ineligible subjects and was deprived of the opportunity to carry out a proper assessment of the impact of protocol deviations or to make decisions, prior to unblinding of the data, with respect to the analysis and reporting of the data and any further amendments to the arrangements for TRUE-AHF [APOC 39(a)-(b)];
(2) The data acquired from TRUE-AHF is not now, and has not at any material time since the database lock (alternatively, the unblinding of the data) been capable of being used reliably or for the purpose for which it was intended, namely to provide reliable Phase III data on the efficacy of Ularitide in relation to the endpoints of the trial that is suitable for the purposes of submission to relevant regulatory authorities and for licensing. [APOC 39(c)]
(3) The results of TRUE-AHF are of little practical or scientific value. [APOC 39(d)]
(4) The money spent on TRUE-AHF has been wasted, in that TRUE-AHF has not been a proper Phase III trial. [APOC 39(e)]
245. As to its loss, Cardiorentis pleaded that by reason of the alleged breaches of contract, it has been deprived of the opportunity to earn revenue on the development of Ularitide. Cardiorentis quantified these losses by reference to the expenditure it incurred on TRUE-AHF, either directly under the GSA or otherwise in reliance on the promised performance by IQVIA of the GSA and CQA. [APOC 40,42]
246. Cardiorentis pleaded, in the alternative, that it had wasted its expenditure on TRUE-AHF in a proportion or an amount to be determined by the Court. [APOC 41]
247. In its pleading, Cardiorentis advanced a further claim against IQVIA RDS, Inc., alleging that IQVIA RDS, Inc. had acted in a manner which constituted unfair or deceptive acts or practices under the North Carolina Unfair and Deceptive Trade Practices Act N.C.G.S. §75-1, by reason of the manner in which it presented the number of ineligible patients to Cardiorentis. Cardiorentis alleged that this was deceptive, and that IQVIA RDS, Inc. conducted itself in a way that was unethical, oppressive, unscrupulous or substantially injurious to consumers.
248. Cardiorentis alleged that this entitled Cardiorentis under North Carolina law to recover from IQVIA RDS, Inc. an award of treble damages, lawyers’ fees and costs. [APOC 43].
249. The final claim advanced by Cardiorentis in its pleadings related to the TRUE-AHF data. Cardiorentis pleaded that:
(1) Under clause 7(a) of the GSA, Cardiorentis is and remains entitled to access to the data and information generated by the trial. [APOC 44]
(2) Further or alternatively, Cardiorentis is entitled pursuant to clause 8 of the GSA to delivery up of all materials, information and all other data owned by it. [APOC 45]
250. In the premises, Cardiorentis sought an injunction that IQVIA do provide access and/or deliver up to Cardiorentis the TRUE-AHF data [APOC 48].
251. In its Defence, IQVIA pleaded that Cardiorentis does not have rights of access to data under clause 7(a) of the GSA, and that Cardiorentis’s rights pursuant to clause 8 of the GSA are subject to Cardiorentis’s compliance with its payment obligations, and therefore subject to IQVIA’s outstanding claims against Cardiorentis. [APOC 68-72]
252. As already stated, IQVIA’s claim in Action CL-2018-000841 was to be treated as a counterclaim to Cardiorentis’s claim.
253. Upon the opening of Cardiorentis’s case at trial, it indicated that it did not pursue its claim under the North Carolina Unfair and Deceptive Trade Practices Act (Cardiorentis, Opening Written Submissions, para. 8).
254. Mr Stanley QC’s opening oral submissions caused IQVIA concerns about whether Cardiorentis would seek to put forward a case of allegations of specific breaches and inadequacies in the conduct of the trial by IQVIA which were not pleaded, in support of a case that there were inadequacies in the data which were not identified in the pleadings. This led to IQVIA serving a Note on this matter, and to Mr Stanley QC confirming, on Day 6, p. 123 that Cardiorentis was neither proposing to amend its APOC, nor to move outside its pleaded case.
The Factual Witnesses called
255. Each side called factual and expert witnesses. Cardiorentis called four factual witnesses: Dr Holzmeister, Dr Mazgareanu, Dr Lustenberger, and Mr Wittwer. IQVIA called 14 factual witnesses: Dr Jeff Spaeder, Tony Owen, Mark Shaw, Claire Raskino, Satish Prabhu, Kenny van Speybroek, Mike Abbs, Lara Queiroz, Matteo Mondelini, Vaida Makunaite-Vilute, Michal Nevo, Aline Ron, Zach Dorman, and Pauljit Hira.
256. All the witnesses were, I was satisfied, attempting to help the court with their best recollection of relevant events. I nevertheless considered Dr Holzmeister’s evidence, in particular, to be, in some respects, lacking in objectivity.
257. More generally, however, this was not a case in which the evidence of the factual witnesses, to the extent that it went beyond what was apparent in the documents, was generally of very great significance. There were a number of reasons for this. Most obviously, several years had passed since the events in question. Secondly, the nature of the subject matter was such that what was important was usually detail which most witnesses could not be expected to remember without reference to documents. Thirdly, and related to the first two, this was not a case in which there were many significant differences of recollection between the witnesses.
258. Both sides commented on the absence of certain witnesses from those called by the other. Given the number of people who were involved in the Study, as well as the points as to the significance of the factual witness evidence which there was and to which I have already referred, I did not, subject to one point, find these criticisms of great importance.
259. That exception is that I did consider that Cardiorentis’s failure to call any of those who had been principally involved in the BDR on its behalf was a matter of significance. While I was prepared to accept that there were particular reasons, about which evidence was given at the trial, as to why it might not have been realistic to have expected Professor Meyer to have given evidence, that did not apply to Dr van Langenberghe, Dr Holcomb or Dr Al-Haboubi. The failure to call any of these witnesses meant that I treated with particular caution any surviving case on the part of Cardiorentis that its team were not properly aware of the number and nature of protocol deviations prior to unblinding; and also Cardiorentis’s claim that there were deficiencies in the BDR.
260. IQVIA also submitted that it was appropriate for me to draw adverse inferences against Cardiorentis’s case from the fact that Dr Packer had not been called as a witness, notwithstanding that he had been named in Cardiorentis’s first Case Management Information Sheet as someone who would be called. While I accept that it would have been helpful to the court to have had evidence from Dr Packer, both because of his role in the study and his eminence as a cardiologist, I concluded that it was not possible to draw any specific inferences, whether adverse to Cardiorentis or not, from the fact of his not having given evidence. Dr Packer has, at different times, said things which can be argued to be favourable to both parties.
261. The parties called expert witnesses to give evidence in relation to three areas: biostatistics, cardiology, and the conduct of clinical trials. On the first, Cardiorentis called Professor Robert Makuch and IQVIA called Professor Jane Hutton; on the second Cardiorentis called Professor Faiez Zannad and IQVIA called Dr Philip Sager; and on the third Cardiorentis called Beth D Harper, and IQVIA called Dr Sager.
262. All experts sought to assist the court. I considered that Dr Sager’s evidence on clinical trial practice was, where they differed, more realistic than Ms Harper’s, various of whose points were not, in any event, pursued by Cardiorentis as criticisms of IQVIA’s performance of the TRUE-AHF trial. I also considered that Professor Hutton’s evidence was, to the extent that they differed, more cogent and more straightforward than Professor Makuch’s. There was a considerable amount of agreement between Dr Sager and Professor Zannad, who are both eminent cardiologists. Dr Sager’s evidence was, however, particularly useful in its breadth and detail.
Agreed facts
263. As I have already said, very many of the facts were not in issue. The parties produced a document in which many facts were agreed, and I have made use of this in the opening section of this judgment. In addition, the parties helpfully agreed, in response to a request which I made, a schedule of the patients randomised into the TRUE-AHF trial, which specified the relevant site and date of randomisation, and whether the patient had an EC3 or IC6 or other eligibility deviation.
264. The parties agreed on a list of issues which arise and which I need to decide. That list is undoubtedly helpful in identifying the points which need resolution. I consider, however, that the order in which the points are listed, which was not the order in which the points were addressed at least by IQVIA in closing, is not that which is most convenient for the purposes of analysis of the claim made.
265. I intend to consider the issues which arise under seven broad headings. I propose first to consider the obligations which IQVIA is said to have been under; and resolve such issues as there are in relation to that. Secondly, I will consider the extent to which it is shown that there were breaches of any of those obligations, and what effect those breaches are shown to have had in relation to the number, nature, incidence of and response to eligibility and, to the extent relevant, other protocol deviations. Thirdly, I will consider the case as to whether there were deficiencies in the BDR process. Fourthly I will consider whether any defects or deficiencies in the study which were the responsibility of IQVIA meant that the data acquired in the trial cannot reliably be used to provide data on the efficacy of ularitide, and/or that the trial was of little practical or scientific value. Fifthly I will consider the legal principles applicable to Cardiorentis’s claim for damages and their application to the facts. Sixthly I will consider Cardiorentis’s claim for an injunction. Seventhly I will consider IQVIA’s counterclaim.
General aspects
266. There were only limited areas of dispute as to the obligations owed by IQVIA under the GSA. The GSA provided for the Services which IQVIA was to perform by Clause 1.0 and Attachment 1. The Services to be provided could be modified by Change Order. The standard to which the Services were to be provided was, by Clause 10(a), that ‘customary in the contract research organisation industry’. The GSA will also have incorporated the implied term under s. 13 Supply of Goods and Services Act 1982, that IQVIA should have carried out the services with reasonable skill and care. If it were the case that the standard ‘customary in the contract research organisation industry’ were lower than a standard of reasonable skill and care, then compliance with the express term would be insufficient. It was not, however, suggested by either party that that was the case, and accordingly, for present purposes, it is not necessary to distinguish between the two standards.
267. The question of what constitutes the standard of reasonable skill and care / the care customary in the contract research organisation industry is one to be determined by the court, assisted by evidence, including expert evidence. The parties informed me that, as far as they were aware, there had been no case decided in this jurisdiction in which the standard of care to be expected of a contract research organisation had been considered and accordingly whatever guidance might have been gained from such a source was unavailable.
268. The Services to be provided to the standard mentioned included the following of particular relevance:
(1) The conduct of Site Initiation Teleconferences (initially anticipated to number 277), and Site Initiation Visits (initially anticipated to number 194 based on there being 194 sites, and with an average time spent at the site of six hours).
(2) Planning and coordinating four two-day investigator meetings, initially anticipated to be for 194 sites.
(3) Developing (while Cardiorentis was to approve) a study reference plan for the project which was to be distributed to the sites, and developing (with Cardiorentis to approve) up to 21 issues of a project-specific newsletter for the sites.
(4) Training the CRAs. This was to include three eight-hour training sessions by IQVIA’s medical advisor.
(5) The conduct of an appropriate number of monitoring visits to sites. It was envisaged at the time of the GSA that these would involve a review of c. 204,000 CRF pages, taking an average of 4 minutes per page. In the event, the number of CRF pages was c. 389,000, at 217 sites, and 3322 site monitoring visits were performed.
(6) IQVIA’s ‘clinical team’ conducting in-house monitoring for an average of one hour per site per month, and its ‘study monitors’ accessing the EDC system to monitor eCRFs, for an average of one hour per site per month. These are under the heading of ‘Clinical Operations Detail’ and I consider are aimed at indicating activities to be performed principally if not exclusively by CRAs.
269. What IQVIA was expected to do in relation to monitoring eligibility deviations was fleshed out and made more specific in the Quality Management Plan, the Monitoring Guidelines and in the PDMP which I have referred to and quoted above. To the extent that these documents contained provisions as to the standard to which services which IQVIA had undertaken to perform were to be provided, I consider that they either constitute Quintiles standard operating procedures, as referred to in clause 10(a) of the GSA, or in any event are an indication of the relevant standard of reasonable skill and care and the standard customary in the contract research organisation industry.
Medical Monitoring
270. One area which requires specific consideration is as to the extent to which IQVIA may have had responsibility for the remote monitoring of the eCRF database. IQVIA pointed to the fact that the GSA provided, in the Scope of Work, under ‘Medical Monitoring Detail’, that IQVIA’s medical advisor would provide only the specified aspects of medical monitoring, and that Cardiorentis would be responsible for all other aspects of medical monitoring. Medical monitoring, as the two experts on clinical trial practice agreed, covers a range of activities, including a review of the electronic database ‘including for inconsistencies, adverse events, protocol deviations, safety issues and prohibited medications etc’, performed by a physician or other qualified individual. The specified aspects of medical monitoring which the Scope of Work referred to as ones which IQVIA would perform did not include the remote review of the electronic database for inconsistencies, protocol deviations or prohibited medications. The provision, in relation to ‘CRA and Site Support’, that Quintiles Medical Advisor would provide medical monitoring activities ‘during normal business hours’ appears clearly to address a type of helpdesk service to address problems and issues which CRAs or Sites might have.
271. That IQVIA’s responsibilities for remote medical monitoring of the database were limited is confirmed by the Medical Monitoring Plan which, as has been set out, provided that Quintiles Medical Advisors were not contracted for remote medical monitoring of eCRFs for data consistency. The reference in that document to ‘Quintiles Medical Advisors will assist [CPMs] in on-going protocol deviation review…’ is, in my judgment, to be regarded as a reference to assistance in the process of reviewing protocol deviations identified by CRAs and which were recorded as such within CTMS and were then listed in the PD Logs. That was the process for ‘Review of Protocol Deviation Listings’ which is laid down in the PDMP in which both CPMs and Medical Advisors had a specified role. It was not a requirement that Medical Advisors should carry out a separate remote review of the database.
272. Similarly, and for completeness, I add that the reference in the GSA’s Scope of Work to In-house Monitoring was not a requirement for the performance of such a review. The In-house monitoring here referred to appears to be intended to be carried out principally if not entirely by CRAs in relation to the records of the sites for which they were responsible, and not to be a requirement for a separate review of the database designed to identify issues which the CRAs did not pick up.
273. I conclude that IQVIA did not undertake a responsibility for remote medical monitoring of the Inform database during the course of the study.
274. There was a further specific issue between the parties as to the extent to which IQVIA was required to perform SDV. The evidence established that SDV is the name given to a process by which a monitor (in the present case typically the CRA) checks the data entered into the eCRF / trial database to confirm that it is accurate, to verify that it aligns with the data in the source documents and that the transfer of information from the source documents and data from the source into the database has been correctly performed.
275. As already set out by reference to the pleadings, Cardiorentis contends that IQVIA had contracted to perform 100% SDV. It refers to the fact that the Scope of Work stated that 100% SDV was assumed for budget purposes; and that while it also stated that a revised plan for SDV would be issued during the course of the study, it never was. It points out further that the Monitoring Guideline referred to CRAs having been ‘asked to conduct 100% SDV for all patients consented to the study’ and had specified data which were to be 100% SDV, which were wide ranging. IQVIA, for its part contended that 100% SDV had not been agreed as a contractual requirement: the GSA had only said that 100% SDV was assumed for budget purposes; and the Monitoring Guideline was not a contractual document.
276. I consider that the contractual requirement was that IQVIA should perform SDV, to the standard customary in the contract research organisation industry, on 100% of the data fields in the eCRF in respect of each patient. 100% was what was specified in the GSA as the basis on which the work was to be budgeted until a revision was put in place, which did not occur. In the circumstances the parties would be reasonably understood to have intended that the only percentage mentioned - 100% - should continue to apply.
277. It is however, germane to mention here certain features of and limitations on the requirement as to SDV.
(1) The first is that the requirement for SDV is not a warranty of the accuracy of 100% of the data in the eCRF for every patient. Even if all datapoints had been verified, there might still be inaccuracies, for instance if the source data were inaccurate.
(2) The second is that SDV is necessarily a moving target. While a patient may be 100% source data verified on one day, if she then acquires more data (for example on the occurrence of an adverse event), the percentage of SDV will fall below 100% for a period. As Cardiorentis points out, however, in relation to eligibility deviations this feature should be of less significance because the source documents required for their verification should have been created at or before the patient was enrolled.
(3) The third is that no specific contractual timeframe was laid down by the GSA for the completion of SDV. The contractual requirement was that it should be conducted in a timely manner, in accordance with the standards customary in the contract research organisation industry.
CQA
278. By the end of the trial, the CQA had assumed a much lesser degree of prominence than it had had at earlier stages in the litigation. There appeared to be no significant dispute as to what the relevant obligations under that agreement were, though there remained an issue as to whether they were breached.
279. The potentially relevant express obligations of IQVIA under the CQA are those set out in clauses 4.4.1, 4.4.2 and 5.2, quoted above.
280. As also set out above, it was pleaded in the APOC that the CQA was governed by the law of North Carolina, and that, judged in accordance with that law, it was an implied term of the CQA that there would be good faith and fair dealing that required neither party to do anything which injured the right of the other to receive the benefits of the agreement. The alleged breach of that implied term was said not to be pursued in paragraph 168 of Cardiorentis’s Opening Skeleton, and it was made clear during the course of the trial that that implied term was not relied upon and no expert evidence was accordingly required in relation to it. While Cardiorentis maintained its contention that the CQA was governed by North Carolina law, and that its express terms fell to be construed in accordance with that law, it accepted (Day 19/4) that there was no material difference between the principles of construction under North Carolina and under English law, and that any issues of North Carolina law fell away.
Were there breaches of contract by IQVIA which caused there to be eligibility deviations?
281. In this section I will consider the allegations of breach of contract on the part of IQVIA, and whether any breaches established are shown to have caused eligibility or, to the limited extent to which they are relevant to the pleaded case, other protocol deviations.
282. I consider together the allegations of breach, and the aspect of factual causation I have identified, for two reasons. In the first place because, given the nature of Cardiorentis’s pleaded case, it is only breaches which had an effect in permitting or not minimising or not alerting Cardiorentis to eligibility (or other protocol) deviations which are relevant. Secondly, because it is convenient, when considering the detail of the alleged breaches to consider at the same time the detail of the extent to which any such breaches can be shown to have affected the incidence of eligibility (or other protocol) deviations.
283. While, on behalf of Cardiorentis, it was said that its case was not one simply of res ipsa loquitur, it was certainly a part of Cardiorentis’s case to suggest that the number of protocol, and in particular, of eligibility deviations which occurred, suggested that something had gone wrong with the study, and was evidence suggesting failures on the part of IQVIA. In the APOC, para. 43(d)(ii) the number of ineligible patients enrolled is called ‘unacceptably high’. In the Amended Reply, para. 34(b), it is alleged that ‘the occurrence of eligibility criteria violations, particularly where such occurrence is repeated and occurred with the frequency that it did in this case, is evidence of failures in training, monitoring and/or reviewing of investigators.’ Dr Holzmeister in his witness statement suggested that the rate he would have expected was in the region of 2-3%, and described the rate of 16.5% as ‘a major problem’. In Cardiorentis’s closing submissions, it was contended that there was ‘an epidemic of eligibility deviations’ (paragraph 72).
284. In examining whether the rate of eligibility deviations can, of itself, be said to be indicative of breach on IQVIA’s part, or, at the least, to call for an explanation by IQVIA, it is right to recall the respective responsibilities of the various parties. It was the Investigators who were responsible for ensuring and verifying that each subject met the eligibility criteria at the time of enrolment. It was they who were present, and they who had to take the decision as to whether the patient should be enrolled. As the experts on Clinical Trial Practice agreed, IQVIA had virtually no control over whether a particular patient was enrolled. Moreover, once a particular patient was enrolled, this could not be ‘undone’, and that patient was part of the study.
285. There was a careful examination before me of what evidence there is as to the rate of eligibility deviations which have been found in other studies, and which might be said to be ‘expected’. It emerged that there are surprisingly limited data, and that those data themselves have to be treated with circumspection, because the number of eligibility deviations is likely to depend in part on the nature of the study. A study of an acute condition, where a decision on enrolment has to be made quickly in challenging conditions in an emergency room (on a ‘sick patient in a chaotic environment’ as Dr Sager put it), is likely to produce a different and higher level of eligibility deviations from that of a study of a chronic condition, where eligibility decisions can be taken more deliberately.
286. It is clear that some eligibility deviations are to be expected in most clinical trials: hence the terms of ICH E9 section 5.2, referred to above. In the Joint Memorandum of the Clinical Trial Experts it was stated as agreed, however, ‘that there is no generally accepted standard as to the number of protocol deviations (including eligibility deviations) which is regarded as acceptable’.
287. There was reference during the evidence to a passage in a book published by Professor Stuart Pocock in 1983 entitled Clinical Trials: A Practical Approach, which said, in part: ‘If the proportion of ineligible patients becomes unduly large, say 10% or more, this may reflect a generally poor standard of trial organisation which needs tightening up. On the other hand it can indicate that the trial’s eligibility criteria are too restrictive, so that investigators are finding that many patients they consider suitable are not actually eligible.’ I accept Dr Sager’s evidence in relation to this, however. The reference to 10% is not tied to any data; it would depend on the type of study; and it must be very questionable as to whether it would be written by Professor Pocock now, especially given the advent, in the interim, of electronic searches of databases, which help to identify additional protocol deviations.
288. Another piece of evidence to which the experts referred was an article by Sweetman & Doig, ‘Failure to report protocol violations in clinical trials: a threat to internal validity’, in Trials (2011). The article reviewed 80 clinical trials published in four major medical journals. The article reported that 32% of studies failed explicitly to report any type of protocol deviation. In relation to enrolment protocol deviations, there was explicit reporting in only 10 of the 80 trials (12.5%). The proportion of participants with such protocol deviations was reported as having a median of 0.8%, with a range of 0.16% to 9.1%.
289. This article is of very limited assistance in the context of the present enquiry. In the first place, only a small proportion of the trials (perhaps as few as 2-3%) will have been acute trials. Secondly, as Dr Sager said, the analysis is likely to be affected by selection bias, in that it is less likely that studies with high rates of protocol deviations will report that fact.
290. A further article, published in August 2021, Historical Benchmarks for Quality Tolerance Limits Parameters in Clinical Trials, by Makowski et al., equally provided limited assistance for present purposes, save to indicate the degree of variability between studies. That article reviewed historical data analyses from 294 clinical trials conducted by seven TransCelerate member companies. The information presented does not break the data down between acute and chronic care trials, and it may well be that little of the data relates to acute trials. Information on eligibility-related protocol deviations was available to the authors only in relation to about 55% of studies considered. The results tabulated relating to ‘All trials’, ie all types and dates of trials which the authors were able to analyse, numbering 141, showed the percentage of trial participants not meeting eligibility criteria with a mean of 6.7%, and a standard deviation of 10.49%. In relation to Phase III studies, the corresponding figures were 7.87% and 11.09%, showing a wide range of results.
291. I accept Dr Sager’s evidence that the best source of comparative data is from the RELAX 1 trial. The FDA’s Clinical Review of that study indicates that 40.1% of the study drug group and 37.1% of the placebo group had protocol deviations; and that 12.9% of randomised patients had eligibility deviations. That percentage is not very different from that of the TRUE-AHF study and, as Dr Sager said, there are reasons for anticipating that the rate of eligibility deviations in the RELAX 1 trial would be lower than for TRUE-AHF. Those reasons included, at least, that RELAX 1 permitted a 16 hour window from first presentation to randomisation, whereas TRUE-AHF had only a 12 hour window with a target of 6 hours from presentation to study drug infusion, and an achieved median time of 6.1 hours. The shorter time period would, as Dr Sager said, mean that people were having to work faster with the attendant enhanced potential for errors in randomisation to occur.
292. The FDA’s Clinical Review of the RELAX 1 trial concluded that ‘the majority of protocol deviations was related to the operational conduct of the study and probably did not impact the interpretability of the efficacy or safety findings.’ More specifically, as Dr Sager, who had been a member of the FDA Panel that reviewed the results of the RELAX 1 trial, pointed out, the FDA did not suggest that the number of eligibility violations impacted efficacy or safety findings either. Nor was there any evidence produced that the RELAX 1 trial was regarded as having been poorly performed, either by the Sponsor, which came to be Novartis, or by the CROs it employed for the purposes of site monitoring for the study.
293. In its Closing Submissions, Cardiorentis accepted ‘that there is no “tipping point” below which eligibility violations are unimportant and above which they are fatal’. I agree with that. More specifically, what I draw from the evidence is that: (1) there is no accepted ‘benchmark’ as to the number of eligibility deviations which may occur in a study of an acute condition such as TRUE-AHF was; (2) regulators, and in particular the FDA, would not adopt an approach which looked simply at the number of eligibility deviations, but would consider the extent to which they affected the reliability of the efficacy or safety data from the study; and (3) that the percentage of eligibility deviations in this case was comparable to that in RELAX 1, and within the range of variability suggested by the results shown in Makowski et al. for Phase III studies. In light of this, I do not consider that there are grounds for considering that the percentage of eligibility deviations in TRUE-AHF constituted, on its own, a ‘red flag’ as to whether the study was competently conducted, or as to whether it would be accepted as an interpretable study by regulators or by the wider scientific community. This emphasises that what will be necessary in this case is to examine in detail the allegations of breach made by Cardiorentis and to see to what extent it is shown that eligibility deviations resulted from such breaches.
Alleged deficiencies in training
294. Cardiorentis made the case that IQVIA was in breach in providing deficient training to Investigators, particularly with regard to compliance with eligibility and entry criteria, and failed to provide adequate further on site training to remedy existing protocol deviations.
295. Insofar as this case was that the training was inadequate in particular respects which led or contributed to the number of eligibility deviations by reason of contravention of EC3 or IC6, it is best addressed in the context of the detailed consideration of those matters below. Wider and more general criticisms were, however, made by Cardiorentis and by Ms Harper of IQVIA’s training, and it is appropriate to consider those at this point.
296. As set out above, an aspect of training for which IQVIA had contractual responsibility was the planning and coordination of Investigator meetings, which provided initial training of Investigators. The documentary record indicates that Cardiorentis and the Executive Committee contributed with IQVIA to the materials used at the Investigator meetings; and national KOLs also made some contributions to Investigator meeting agendas and slides. Dr Holzmeister, Dr van Langenberghe and Professor Meyer between them attended and gave presentations at eight Investigator meetings. Dr Holzmeister did not make contemporary criticisms of the Investigator meetings, and he did not recall any complaints about the quality of training at an Investigator meeting which he attended. Ms Harper indicated in her oral evidence that she did not have specific criticisms of the conduct of the Investigator meetings. Subject to the point that I will consider separately the specific allegations in relation to EC3 and IC6 deviations, I conclude that no case of breach by IQVIA in relation to the conduct or contents of Investigator meetings has been established.
297. In relation to site initiation visits, Ms Harper criticised the materials which IQVIA had produced for these. She gave evidence that in her opinion the Powerpoint presentations ‘simply copied and pasted the I/E [inclusion/exclusion] criteria … without any additional elaboration, commentary, case study examples and without other means to test or validate the site’s understanding of the criteria’, and that the only ‘job aid’ was a pocket-sized card with the I/E criteria listed. The difficulty with this evidence is that it is plain, and Dr Holzmeister did not dispute, that in a presentation it will have been the key points which were put on the slides, and there will have been oral elaboration. As there was no examination of the specific content of any particular site initiation visit, no case was made out that, taking both the slides, the card, and the oral elaboration, the training was inadequate. In relation to a site initiation visit which Dr Holzmeister had himself attended, at Basel, he had not had complaints as to the way it was conducted, but had regarded it as a good meeting. I was unpersuaded that Dr Holzmeister would not have been able to detect whether the training at the visit was inadequate, and was in no doubt that had he considered it to be inadequate he would have said so. Furthermore, Dr Holzmeister did not recall anyone else complaining at the time about the standard of training at site initiation visits.
298. No inference can be drawn that the training at site initiation visits was inadequate from the simple fact that eligibility deviations occurred, not least because in the great majority of cases the patients enrolled were eligible. It is apparent that sites were able to get enrolment right. This tends to suggest that the reason for eligibility deviations was not because of any general problem with the training materials which were used, but was due either to particular failures in specific visits, a case which Cardiorentis has not established, or to failures on the part of some sites, or some personnel at some sites, to absorb the training which they were given.
299. Ms Harper made similar criticisms of the materials made available by IQVIA for training its CRAs as she did of the materials made available for training the sites. Her opinion was that it was not adequate to have Powerpoint presentations which simply copied out the inclusion and exclusion criteria; and the five question test which CRAs had to take was not sufficient. She acknowledged that one of the steps she had recommended - the provision to CRAs and sites of ‘pocket protocols’ - had been taken.
300. As with the criticisms of the site initiation visits, I was not persuaded that it had been shown that IQVIA was at fault. Here again, the Powerpoints will not have been the totality of what was said in training the CRAs, and there was no detailed examination of how it was said that the training provided on particular occasions fell short. Dr Holzmeister had attended a CRA training session for European-based CRAs in Paris in November 2012 and a WebEx call with CRAs based in the USA, also in the second half of 2012. Dr Holzmeister remembered that the Paris meeting had been a good meeting. He had been sent the Protocol training slides both before that meeting, and after it, when they had been updated to reflect feedback received from CRAs. He did not make any criticisms contemporaneously as to the quality of the training of CRAs which he had witnessed or of the content or use made of the presentations. This is cogent contemporary evidence that the process of training CRAs was considered adequate.
301. Again, moreover, the fact that eligibility deviations occurred does not of itself show that the process of training CRAs was defective. The large majority of patients enrolled were eligible. To the extent that the enrolment of an eligible or ineligible patient can be attributed to the training of the CRAs who trained the sites, this suggests that the training was sufficient to allow CRAs to train sites properly. Once again, there is the clear possibility that any defects were the result of the variable responses of the sites to the training they had received. Cardiorentis bears the burden of proving the breaches by IQVIA for which it contends, and it has not shown that that possibility is less likely than that any failures were the result of inadequate training of CRAs.
302. Cardiorentis’s allegations as to failures of proper monitoring are central to its case on breach. Much of the detail of its case on monitoring is part of, and is best considered in the context of, its case in relation to EC3 and IC6 violations, and certain other protocol violations. Again, however, there are certain wider or more general aspects of its case which it is convenient to consider first.
303. There was no dispute that effective monitoring by CRAs was very important to there being a successful trial. Ms Queiroz and Mr Mondellini accepted that it was critical to the project. The important role of monitoring visits in terms of identifying protocol violations is recognised in ICH-GCP E6, and the Quality Management Plan.
304. Cardiorentis complains that the monitoring process can be seen to have gone seriously wrong, by reason, at least, of the following facts: (i) that there were 365 eligibility violations; (ii) that of those, 165 subjects (with 175 instances of eligibility violations) were not detected by CRAs, but were identified by BIOS searches; (iii) that in the case of many of the eligibility violations which were identified by CRAs, this occurred only at the second, or a later, site monitoring visit after the randomisation of the ineligible patient.
305. Insofar as Cardiorentis relied on the suggestion that it ‘speaks for itself’ that there must have been failures by CRAs to monitor properly and promptly, by reason of these matters, I do not consider that such a case can be successfully made in this case. It was apparent from the evidence that there may have been many reasons why there might have been delays or difficulties in the identification of eligibility or other protocol deviations, and in the elimination of such deviations, which were not the fault of the CRA. These included that:
(1) The Principal Investigator, or another key staff member, was not (or would not make him/herself) available to speak to the CRA. Ms Queiroz said this:
‘It may be … that the principal investigator was not available there, or the key staff member that he needed to speak to wasn’t there. … It may have been that the site staff didn’t make time to speak to the CRA, because that happens a lot. Sometimes the CRAs go to the site, they are provided with the documents and nobody or some key team members from the site do not appear to speak to them…’ (Day 11/134)
(2) The necessary source documentation may not have been made available to the CRA when (s)he attended. Ms Queiroz gave evidence as to the problems which could occur in a particular case as including ‘not having the source documents available’ (Day 11/142), and Ms Makunaite-Vilute of a site which ‘did not provide sufficient source documents to us for review’ (Day 14/16).
(3) Some data may not have been promptly entered into the eCRF by busy Investigators. Mr Mondellini explained delays in getting data clean and into the system as, at least in part, due to ‘the investigators … working in an emergency environment, so they have not a lot of time for entering the data into the database’ (Day 13/141).
(4) Action items may have been in the hands of the Investigators. Ms Makunaite-Vilute gave evidence that an item had been open for a considerable time in relation to one of the patients enrolled at Spanish site 2505 because a note signed by the Principal Investigator was awaited (Day 14/20-21).
(5) A problem at a site may have been one which was raised with the site by a CRA and had led to retraining by the site, but where the site nevertheless experienced ongoing difficulties in ensuring compliance with the Protocol. This is what appears to have occurred in relation to dosing discrepancies at site 1707 in the Netherlands, which was the subject of an email chain of 11-12 June 2015 about which Ms Makunaite-Vilute gave evidence (Day 14/24-28).
(6) If a large number of patients were enrolled in a short period of time, it may not have been possible for a CRA to check them all in one monitoring visit. Ms Makunaite-Vilute gave evidence to that effect (Day 14/31).
306. It is also pertinent to record that Cardiorentis made no case that IQVIA did not perform in accordance with its contractual obligations as to the number of monitoring visits to be undertaken, or as to the number of hours to be dedicated to monitoring by CRAs. There was thus no general case that IQVIA failed to devote the contracted for level of monitoring activity.
307. In the circumstances, Cardiorentis can succeed in showing a breach in relation to monitoring activities to the extent that a specific case is made that particular eligibility or other protocol deviations were caused by particular failings of monitoring and follow up. I turn to consider the more specific allegations which Cardiorentis made.
308. An important aspect of Cardiorentis’s case on breach was its allegations that it was IQVIA’s failings which had led to the number of patients enrolled who were ineligible by reason of violation of EC3. That criterion is set out above, but it is convenient to repeat it here:
‘3) Treatment with levosimendan, milrinone, or any other phosphodiesterase inhibitor within 7 days before randomization.’
309. In the SCI analysis 124 patients were identified as having been included in the study who were ineligible by reason of breaches of EC3. In the agreed schedule produced during the course of the trial, 126 patients are classified as having been ineligible by reason of violations of EC3. Of the 124 who were in the SCI analysis, 72 were from sites in Serbia, and 46 at site 2006 there.
310. As was set out by Dr Sager, levosimendan and milrinone are phosphodiesterase inhibitors. Those two drugs, and amrinone, are used to treat ADHF patients who are very sick. They are vasodilators, and positive inotropes (ie they help the heart to contract more efficiently). ‘Other phosphodiesterase inhibitors’ is a much wider category of drugs. There are many types of phosphodiesterase enzymes which have physiologic activity in different tissues and exert a wide variety of different functions. PDE inhibitors are accordingly a large class of drugs which are used for many different indications, many having no role in treating cardiac diseases. Caffeine, for example, has PDE inhibitory effects, as does, at least at high doses, theobromine which is found in chocolate and tea, and Viagra.
The facts relevant to EC3 deviations
311. The facts most relevant to Cardiorentis’s case as to EC3 can be summarised in the following manner. In giving this summary, I do not intend at present to deal with IQVIA’s case that in relying on these matters Cardiorentis was, in some respects, going outside the case which it was entitled to run on the pleadings.
312. At the outset and in the early part of the study, no itemised list was supplied to sites of prohibited medications, treatment with which meant that a patient should not be included in the study. There was, in particular, no list of ‘PDE inhibitors’.
313. At a site initiation visit at one site (0401) in Kansas, USA, on 17 January 2013, one of the issues discussed between the CRA, Jane Notaro, and the Investigators at the site was that the site ‘requested a list of the medications excluded’ by EC3. In an email from the CRA to Amy Finn, her CPM, and Veronica Kohl, Clinical Project Support Specialist at IQVIA, on 18 January 2013, Ms Notaro recorded that many questions had been discussed at the site initiation visit, and mentioned a considerable number, one of which was ‘Exclusion Criteria #3 lists concomitant medications excluded - The site asked for a cheat sheet with all meds falling into these categories.’ Ms Kohl added the comment ‘Ana do you agree to this?’
314. In the follow up letter to the site after the site initiation visit, which was dated 30 January 2013, amongst a number of other points, the site’s request was said to have been ‘forwarded to the Sponsor/other resources for follow-up’ and to be ‘pending’. The point then appeared in successive iterations of IQVIA’s Action Item Tracker, which was automatically populated from ‘action items’ in the monitoring visit reports, as a ‘minor’ issue, but one which was ‘overdue’. It is not clear whether the point was raised with Cardiorentis. IQVIA contends that it must have been and that accounts for the fact that the letter of 30 January 2013 was in Cardiorentis’s possession and was disclosed by it in this litigation.
315. At the first site monitoring visit to site 0401, which occurred on 22 January 2014, just after the first patient was enrolled to the trial at that site on 14 January 2014, there was a discussion about a large number of issues. One of these was in relation to the site’s request in relation to EC3 medications. The Resolution is recorded as ‘A list was not provided by Quintiles. The CRA did discuss this criteria with the SC and PI and they are aware of the exclusionary medications.’
316. Site 0401 at Kansas did not enrol any patients with EC3 violations during the course of the study.
317. After a site initiation visit at site 1906 in Romania on 7 June 2013, the follow up letter recorded that one question which had been raised by the site’s representatives was ‘Is eligible a patient treated with Theophylline?’. This was said to have been ‘forwarded to the Sponsor/other resources for follow-up’, and the letter said that the CRA would ‘contact [the site] via email/phone once a response is provided’. No record of what occurred was produced during the trial. This site did not enrol any patients with EC3 deviations.
318. After a site monitoring visit at site 1803 in Poland, which occurred on 21-22 July 2014, it was recorded in the Report, in relation to one patient who had been enrolled on 7 October 2013, that ‘Exclusion criterion 3 was met, subject took Theophylline 7 days prior to randomization.’ The action taken was described as ‘CRA retrained SI [senior Investigator] on exclusionary concomitant medications as per exclusion criterion 3’ and recorded that no further action was still required. No further EC3 deviations occurred at this site.
319. A similar pattern occurred at another site in Poland, site 1806. After a site monitoring visit on 24 July 2014, it was recorded in the Report that a patient, enrolled on 29 November 2013, had met EC3 because ‘theophillinum (sic) administered before randomization.’ The action taken was described as ‘Dr Lewandowski was on annual leave and shall be retrained during next SMV. Training to be documented on training log.’ No other EC3 deviations occurred at site 1806 apart from the one patient to whom this relates.
320. The extent of the issue relating to EC3 violations was brought to light following the Data Management audit of IQVIA, conducted at Illkirch by Dr Lustenberger and Dr Rother on behalf of Cardiorentis on 11-12 June 2014. The overall conclusions of the auditors included that ‘the Data Management team [at IQVIA] is well organised and SOPs are in place’; that ‘No finding was considered critical’; and that ‘the auditors are convinced that the Data Management team under the leadership of Marie Golema should be able to resolve all DM related issues and deficiencies within a reasonable amount of time.’
321. One ‘Minor finding’ of the auditors was expressed as ‘DM Quality Plan: The medical review of listings of prohibited medication was defined as not applicable. But it is applicable for this study and it is unclear how it is currently done.’ The Recommendation was expressed as ‘During the audit, it was not checked whether such medical review is stipulated, but in the auditors’ view such review is necessary for this study. This should be changed accordingly. A description of how the medical review is done should be given.’
322. On 11 August 2014, Ms Golema sent an email to Sherry Popiolek, Matteo Mondellini and others, enclosing a draft Data Management CAPA which included this item. There was then a series of emails within IQVIA’s team which debated the extent of IQVIA’s responsibility in this area. Ms Golema considered how the prohibited concomitant medications which were not permitted during the first 72 hours following the start of infusion, which included levosimendan, milrinone and other PDE inhibitors, might be identified. She said that medical input was required regarding a list of PDE inhibitors; and she attached a Standardised Drug Grouping listing of all PDE inhibitors identified by the Uppsala Monitoring Centre.
323. On 14 August 2014, Ms Golema sent an email to, inter alios, Veronique Mahaux, Rafal Ziecina and Mr Mondellini, which said in part:
‘Regarding the fact that this item has been highlighted as a finding for DM, I have checked how these deviations are usually tracked for other studies/Sponsors.
For most of these studies, CRAs/Mas [medical advisors] are responsible for the identification of protocol deviations (including the use of prohibited medications).
The identification of these deviations is based on SDV made by the CRA and the review of eCRFs/ patient profiles by the Mas without any involvement of DM.
DM would specify and program protocol deviations checks if the identification of these deviations is included in DM SoW [scope of work] and managed within the eCRF (not CTMS).
As we could have unreported deviations, I agree DM needs to be involved in the identification of this deviation by programming an automated check.
However, this won’t replace a medical review and similar issues can exist regarding other type of protocol deviations.’
In the same email Ms Golema said that, once the list of prohibited medications was finalised, it would require a week to programme an electronic search for deviations involving the prohibited medications within eCRF.
324. By 27 August 2014 Ms Golema had conducted the electronic search of eCRF. This had identified 150 patients who had used prohibited medications (before or after initiation of the study drug). On 28 August 2014, Ms Golema sent to Mr Mondellini and CPMs lists of patients identified to have taken prohibited medications. She mentioned the matter again on a data cleaning call on 25 September 2014 involving Mr Meller, Ms Queiroz and Ms Finn, and resent the lists of patients, which identified also the site and the prohibited medication concerned. This led to action from Ms Queiroz and others. She sent to the CRAs in Latin America a list of the protocol deviations found by Ms Golema relating to Latin American countries. On 3 October 2014 she also included in the weekly newsletter to all CRAs a list of PDE inhibitors approved by IQVIA’s medical monitors. It was based on the same SDG listing that Ms Golema had earlier used. It ran to some 1916 entries, and contained aminophylline and theophylline (as well as caffeine, Beecham’s hot lemon cold remedy, Lucozade and many others).
325. The list of prohibited medications was sent to sites on 15 October 2014 in the regular newsletter. This was sent out by Cardiorentis, and accordingly someone at Cardiorentis was at least aware by this date that the list had been identified and was being sent out, but there is no indication that it had at this stage been told about the data management searches which Ms Golema had performed. On 22 October 2014 Teiba Al-Haboubi of Cardiorentis asked Ms Golema whether there was an ‘automatic query fired’ when a site reported a prohibited medication. Ms Golema replied that no automated check was in place; and referred to the listing of prohibited medications which had recently been sent to CRAs.
326. On 21 November 2014, Jelena Svatok, a CRA based in Belgrade, Serbia, sent to Michal Meller, Rafal Ziecina and others an email which referred to the newsletter which had forwarded the prohibited medication list containing aminophylline. Ms Svatok wrote:
‘In accordance with our National Register of Drug, Aminophylline is classified in ksantin group of drug with primary bronchodilatative activity. Phosphodiesterase inhibition is dose dependent (Aminophylline dose, which is needed for PDE inhibition is significantly higher than Aminophylline dose for therapeutic bronchodilatative activity). …
Please find below mail from our National coordinator, Professor Petar Seferovic:
Dear All,
We were recently (two weeks ago) notified that Aminophylline is listed as prohibited medication in the TRUE AHF study. In all versions of protocols, Levosimendan and Milrionone (sic), but not Aminophylline were highlighted by name as PDE3 inhibitors.
According to Serbian pharmacological registry, Aminophylline is classified as brochodilatator (sic), and is widely used ONLY for that specific indication.
The effects of the drug includes central (brain), bronchial and peripheral vascular effects, as well as mild diuretic properties.
Per protocol, all drugs (including Aminophylline) can be used after randomization, as per physicians discretion.
It came as a surprise that this issue arise after almost two years after the beginning of the study, probably because Aminophylline was not considered a specific PDE 3 inhibitor….’
327. After this was circulated within IQVIA, on 27 November 2014 Ms Queiroz identified, from Ms Golema’s lists, that 46 patients had protocol deviations by reason of having taken aminophylline. On the same date Rafal Ziecina emailed Cardiorentis (including Dr van Langenberghe, Professor Meyer and Dr Holzmeister) forwarding Professor Seferovic’s comments. Dr van Langenberghe forwarded the email to Professor Meyer commenting that he thought that the issue was an interesting one, and suggesting that Professor Meyer should handle it with Professor Seferovic, which Professor Meyer then agreed to do.
328. On 4 December 2014, Oie Podra, a CRA in Estonia, reported to Ms Ron that
‘Dr Põder - PI at site 0701 was disagree with the PD and not willing to change the data.
He explained that in the provided list of phosphodiesterase inhibitors are also medication like coffein (sic) or ephedrine which does not have a stimulatory effect on the actual heart action. He was concerned about way of the interpretation as 4 mcg/kg/min dobutamin is allowed to use but aminophylline is small doses are prohibited - however the dobutamin effect cannot be compare (sic) with effect of aminophyllin…’
329. On 4 February 2015, Professor Meyer emailed Rafal Ziecina and Jason Turner, asking to discuss the process for the medical review for prohibited medications of patients enrolled on the trial, and asking for an estimate of how many patients had been identified as having received prohibited medications and how they were distributed by site and country. On the following day, Professor Meyer elaborated on the checks which he considered might be made, including an exercise of database extraction and review. On 6 February 2015 Professor Meyer wrote that it was important to understand how many patients might be affected in total. On 6 March 2015, Ms Queiroz sent to Cardiorentis (including Dr van Langenberghe, Dr Al-Taboubi and Professor Meyer) and to many of those concerned within IQVIA, the results of the process of data extraction and review that had been conducted up to then. It was reported that the number of patients identified as potentially having breached EC3 was 44 potential violators and 4 confirmed (in CTMS); while potentially 194 patients might have received a treatment with a prohibited medication in the 72 hours after the start of infusion with the study drug.
330. On 15 June 2015, Ms Queiroz reported to Dr Al-Haboubi that there had now been identified 50 eligibility violations as a result of prohibited medications, 26 of which had now been reported in the CTMS system. There were periodic subsequent updates, the final one of 15 January 2016 referring to 62 potential eligibility violators. Subsequently in the final PD Spreadsheet which embodied the result of BIOS searches, the number of EC3 violations was identified as 102, the difference from the information of 15 January 2016 being an extra 40 cases at site 2006 in Serbia.
331. Against the background of these facts, and without yet addressing issues of fault to which I will return, I identify the following combination of matters as having given rise to a large number of the EC3 violations:
(1) That aminophylline, and theophylline which is a very similar drug, are drugs usually administered orally to treat pulmonary conditions. They are not widely prescribed now in North America or in Western Europe. (Sager, first report para. 225) But in some countries they are used to treat some of the symptoms of ADHF.
(2) Aminophylline and theophylline may have little or no PDE inhibitory effect at therapeutic doses. (Cardiologist experts Joint Memorandum, paras 25-26)
(3) Partly as a result of that, in some countries aminophylline / theophylline are not classified or primarily regarded as PDE inhibitors. This included in Serbia, where aminophylline was classified in the xanthine group, with primary bronchodilation activity. ALIMS, the Medicines and Medical Devices Agency of Serbia still classifies ‘Aminofilin’ as a bronchodilator and not a PDE inhibitor, and the Serbian SMC (summary of the characteristics of the medicine) makes no reference to Aminophylline being a PDE inhibitor (Sager Supplemental Report, para. 91).
(4) Moreover, in some such countries, even senior and eminent cardiologists were unaware that the local classification differed from other classifications.
(5) This resulted in Investigators in some countries, of which Serbia was the most significant example, enrolling patients who had been treated with aminophylline, without appreciating that it should be regarded as a PDE inhibitor for the purposes of the Protocol, including EC3.
Cardiorentis’s case
332. Cardiorentis’s case is that such enrolment would not have occurred, or not have occurred to the extent it did, but for breaches by IQVIA of its contractual obligations. As I understood it, this case had five aspects.
333. The first was that IQVIA should have ensured that CRAs and sites were given lists of prohibited medications at the outset of the study. I do not consider that this is well-founded. Dr Sager’s evidence was that it would ordinarily be the Sponsor’s decision as to whether such a list was necessary; and that he was not surprised that there was no such list in the present case. Ms Harper agreed that in some cases there was such a list, and in some cases not, though she said that in a majority of cases there was a list.
334. In the present case it appears clear that a list was not thought to be necessary. All those involved in the study knew that there was no such list. That includes the principal individuals at Cardiorentis, the Steering Committee, the Executive Committee, co-monitors, auditors and regulatory inspectors, as well as IQVIA. Dr Holzmeister’s evidence, in particular, was to the effect that he had not thought a list necessary (Day 2/110-111). I do not consider that the view that a list was not necessary to have been an unreasonable one.
335. I should add that insofar as any case was made that a list should have been considered necessary by IQVIA because there should have been an appreciation of the potential difficulties which might arise from differential classification of PDE inhibitors in different countries, I do not accept it. I consider it to be clear that this was a problem which was not reasonably to be anticipated by a CRO in IQVIA’s position. I accept Dr Sager’s evidence in his Supplemental Report at para. 91 and at Day 19/56.
336. This conclusion is supported by a consideration of the position of Professor Seferovic. Professor Seferovic was a distinguished cardiologist in Serbia. He had been President of the Heart Failure Association of the European Society of Cardiology. He had also been an investigator on the SIRIUS II Phase II trial in which ‘phosphodiesterase inhibitors’ were listed as prohibited medications. He was a member of the Steering Committee for the TRUE-AHF trial. He was very well qualified to identify the potential problem of ‘PDE inhibitors’ not being understood as embracing certain drugs which might be in use in certain Eastern European countries. Yet he did not; and as he wrote in November 2014 he was surprised that the issue had now arisen.
337. The second aspect of Cardiorentis’s case was that, even if it had reasonably been thought unnecessary to have a list at the outset, one should have been provided when relevant questions were raised with IQVIA by others. This case relies heavily on the request made by Kansas site 0401. But in my view it is significant that that was simply a request made by one site, at a site initiation visit. It was not made in the context of any difficulty which had been experienced in giving effect to EC3, as the site had not enrolled any patients at the time of the request. The request was not related to the issue of whether aminophylline did or did not count. It may be that the response, which was not to provide a list, was approved by Cardiorentis, which was in possession of the follow up letter of January 2013, but there is no other evidence of this and I am unable to conclude that Cardiorentis did approve it. Whether the decision was that of Cardiorentis or of IQVIA, however, I consider that if it was reasonable not to provide a list in the first place, it was reasonable not to provide one when a single site asked for one without having a specific reason for needing it. The fact that there would not be a list appears to have been accepted by the site, and gave rise to no problems at that site.
338. Cardiorentis also sought to place reliance on what happened at Romanian site 1906 in relation to the query as to theophylline, and Polish sites 1803 and 1806 in relation to EC3 deviations, which are summarised above. These were matters which were not contained in Cardiorentis’s pleadings, nor in the expert evidence served on its behalf. Doubtless this contributed to there being very little evidence available as to what had occurred. Furthermore, in light of the absence of pleaded complaints, I did not consider that it was open to Cardiorentis to make criticisms of specific failings on the part of the CRAs or other IQVIA staff involved in these matters, because no proper notice had been given of such allegations.
339. In relation to what happened at the site in Romania, and taking into account that there was no pleaded complaint, I do not consider that there is any basis for concluding that the CRA to whom the query as to theophylline was addressed did not properly address it, perhaps in conjunction with a medical adviser. There may have been a response to the site by phone, as envisaged by the follow up letter. What is known is that there were no EC3 deviations at this site. Nor do I consider that it can be said that a proper follow up of this one query, in the absence of any reason why those involved should have perceived there to be a more general issue as to the ambit of the PDE inhibitors referred to in the Protocol, should or would have led to the production of a list of prohibited medications circulated to CRAs and sites.
340. In relation to the two sites in Poland, Cardiorentis sought to criticise the length of time which elapsed between the randomisation of the two patients concerned and their identification. Again, I considered that these criticisms constituted or involved unpleaded criticisms of the conduct of individual CRAs; and I did not consider that IQVIA’s witnesses could fairly be expected to deal with such detailed matters raised for the first time in cross-examination. I do not consider that it can be concluded that these delays were the result of fault on the part of IQVIA, and, indeed, in relation to Site 1803, a comment was entered within the subsequent PD logs that there had been late reporting ‘due to poor site performance’, suggesting that it was considered that the site was at fault.
341. The response of the CRAs, once the eligibility deviations had been identified, in retraining the sites, appears to have been appropriate. In any event, the two eligibility deviations due to the use of theophylline were identified in July 2014. This was only shortly before Ms Golema addressed the issue of PDE inhibitors in the context of the Data Management CAPA. Even had the identification of these protocol deviations called for different action and further escalation, which I do not find, this would have been unlikely to have led to a list of prohibited medications being sent out much earlier than it in fact was.
342. The third strand was intimated in Ms Harper’s first expert report, namely that IQVIA should have provided for an automated edit check or auto query in the eCRF to identify prohibited medications when they were first entered on the CONMEDS page. I accept IQVIA’s case, however, that there was no contractual requirement on it to provide this. It was not stipulated for, and it appears that Cardiorentis appreciated that it was not being done (see Dr Lustenberger’s Witness Statement, para. 60). The suggestion that IQVIA was at fault in failing to provide such an automated check was not pursued in cross-examination of IQVIA’s witnesses or mentioned in Cardiorentis’s Closing Submissions and, to the extent not abandoned, I reject it.
343. The fourth strand was Ms Harper’s suggestion that the CRAs, including in Serbia, ought to have identified that aminophylline was a PDE inhibitor when they came across a reference to this drug in any patient’s records (Day 18/71-73). This case was again one which implied criticisms of what particular CRAs did, when there was no particularised case as to this, and little material to go on. I considered, in any event, that this case was an unrealistic one. The primary responsibility for assessing eligibility lay with the Investigators, who were experts in their fields. Had a CRA in, to take the most important example, Serbia, considered that it was necessary to raise a question about aminophylline, and whether it was a prohibited medicine, their first port of call would be likely to be the Investigator, as Ms Harper herself said. It appears clear that had Investigators in Serbia been asked as to whether aminophylline was a PDE inhibitor, they would have said that, at therapeutic doses, it was not. I do not accept that a CRA should necessarily have questioned such an answer or taken the matter up with others.
344. The fifth strand was that there were delays in IQVIA’s dealing with and reporting the issue once it had been identified. There seemed to me to be force in Cardiorentis’s point that there was some delay, for which there was no good reason, in IQVIA taking steps to address the issue. Specifically I consider that there was no good reason for the delay of about a month after Ms Golema had the results of her data management searches on 27 August 2014 until the data cleaning meeting on 25 September 2014 which, as Cardiorentis accepts, prompted ‘swift action’ which ‘in large measure’ resolved the issue. During that period of delay, however, only two patients were randomised who had EC3 violations. Accordingly this delay had very limited causative significance.
345. There also seemed to me to be force in Cardiorentis’s complaint that the fact and results of Ms Golema’s data management searches which had occurred by the end of August 2014 ought to have been reported to Cardiorentis earlier than they were. Again, however, it is difficult to attribute to this delay any significant consequences. The list of PDE inhibitors was sent out to CRAs and sites before the results of Ms Golema’s searches were communicated to Cardiorentis. I do not consider that it has been shown that, had Cardiorentis been informed earlier, significantly more would have been done to avoid the problem than was in fact done, which involved the sending out of such lists with reminders to CRAs and sites that the matter must be considered. In any event, the number of patients enrolled after mid-September 2014 who had EC3 violations was not very large (about 18), and some of those (eg EC3 violations in Argentina, Italy and Spain) may have involved different issues from that concerning aminophylline, and might not have been prevented by any response to the emergence of that issue.
346. In summary, therefore, I do not find Cardiorentis’s case that the number of EC3 violations which occurred was due to fault on the part of IQVIA to have been made out. In relation to the main issue which has been canvassed, and which greatly contributed to the number of EC3 violations, namely that aminophylline (and theophylline) were not regarded in some countries as PDE inhibitors, I agree with Dr Sager’s evidence (Day 19/56-57). This was not an issue which IQVIA or any reasonable CRO would have expected or should have anticipated; and once it had been identified there were, albeit after an avoidable delay of about a month, active steps taken to resolve it.
347. The second category of eligibility violations which Cardiorentis identified and particularly relied upon as being largely caused by and indicative of breach on the part of IQVIA was IC6 violations. Again, it is helpful to recall its terms, which were that, to be eligible for randomisation, a patient should have:
‘6) Persisting dyspnea at rest despite standard background therapy for ADHF (as determined by the Investigator) which must include IV furosemide (or equivalent diuretic) at ≥40 mg (or its equivalent) at any time after start of emergency services (ambulance, emergency department, or hospital). At the time of randomization, the patient must still be symptomatic. In addition, the patient should not have received an IV bolus of a diuretic for at least 2 h prior to randomization, and the infusion rates of all ongoing IV infusions [of medication to treat HF] must not have been increased or decreased for at least 2 h prior to randomization.’
348. In the SCI analysis, 189 patients were included in the group of patients who were ineligible by reason of violation of IC6. In the Agreed Schedule of patients included in the trial with eligibility deviations, 192 patients are identified as having had IC6 violations. The IC6 violations shown in the Agreed Schedule occurred at some 72 sites in 19 countries.
The facts relevant to IC6 deviations
349. Once again, it is convenient to summarise the principal facts relating to IC6 violations to which attention was given during the trial, as I find them to have been, before considering Cardiorentis’s case as to breach in relation to those violations.
350. On 2 August 2012, as part of the Project Risk log circulated by Ana Gonzalez, one ‘study design risk’ was identified as:
‘Once all eligibility criteria are met, and before randomization, the subject must still be symptomatic to be eligible for the study. In addition, HF-related IV medications must be unchanged over the last 2 hours. There is a risk the final evaluation prior randomization being missed.’
351. By 1 June 2013, some 17 patients had been randomised in violation of IC6, but only 3 had been reported in PD Logs by that date. By 1 September 2013, there had been some 42 violations of IC6 but only 7 had been reported; and by 31 October 2013 the figures were 61 and 8.
352. As part of its remote medical monitoring, IMR included an entry in its ‘Remote Monitoring Issues Log’ dated 14 October 2013. This entry, dated 26 September 2013, was: ‘Inclusion criterion #6 (iv bolus and change of iv meds 2 hrs prior to random) misunderstood/neglected.’ The Follow Up comment was ‘Include topic in Newsletter / routine monitoring to detect and discuss with site - included in reminder to CRA.’ The column as to closure of this issue was marked Y, ie ‘Yes’. It does not appear that this topic was included in a newsletter to sites or CRAs at the time.
353. At a site visit to a site in Rome (1605) on 18 November 2013, the CRA (Ms Landolfi) identified 4 eligibility violations by reason of a change in IV therapy within the two hours prior to randomisation. The issue was that the CRA had identified that the nitroglycerin infusion for a patient had been interrupted one hour before randomisation. The site had considered that IC6 referred only to diuretic infusions, and not to all other infusion therapies. The site visit report recorded that the site had been retrained on this aspect. According to the Agreed Schedule, only one further patient was subsequently (much later) randomised at site 1605 in contravention of IC6.
354. Ms Landolfi’s internal report of what had occurred at her site visit to site 1605 provoked some internal discussion within IQVIA. Dr Eva Cojocaru, on 5 December 2013, expressed the view to Ms Aguirre and Ms Landolfi, that IC6, which she quoted in what was its terms prior to Protocol version 03, did not refer to all medications. At much the same time, in the context of a query arising from a site in Poland (1807), Dr Holzmeister was consulted on the interpretation of IC6, and in the light of that consultation, Dr Mahaux wrote on 17 December 2013, including to Michal Meller and Eva Cojocaru:
‘Although the protocol states that any changes in on-going infusion within the 2 hours prior randomization would correspond to an exclusion criteria (or that at least the randomization will be postponed), the intent of the eligibility criteria is to ensure the subject was stable and on-going treatments well defined / stable at the time of randomization.
· It is mainly referring to diuretics and vasoactive substances or vasopressors.
· A change in a KCI perfusion would NOT be considered a violation.
· Nonetheless, the administration of albumin would be considered as a deviation and a contra-indication to randomization because of osmotic repercussions.’ (emphasis in original)
355. Dr Cojocaru understood this guidance as being applicable to the query arising from the Rome site visit as well.
356. On 26 August 2014, Dr Rafal Ziecina requested IQVIA’s Data Management team to run a listing to identify potential deviations in relation to whether ‘infusion rates of all ongoing IV infusions must not have been increased or decreased for at least 2 h prior to randomization’. Such a listing does not appear to have been created at this point.
357. By the end of August 2014, there had been some 128 patients enrolled with IC6 violations, of which 25 had been reported in PD Logs. Many IC6 violations were identified for the first time in the BIOS listings.
358. There is no doubt, and it was not in issue, that IC6 was a complex criterion, involving a number of sub-criteria, and multiple repeated assessments over a short term. It was the type of criterion which sites were likely, at least sometimes, to misapply. Cardiorentis’s case, however, was that the number of IC6 violations was IQVIA’s fault.
359. Again, as with EC3, I consider it helpful to consider the strands of Cardiorentis’s case in this regard separately. I identify three.
360. The first strand is related to Cardiorentis’s more general case, which I have already considered, as to deficiencies in training of CRAs and sites. In relation specifically to IC6, Cardiorentis contended that, because it was a complex criterion, special attention needed to be paid to it, and it was not enough simply to put it on a presentation slide. Here again, however, it is apparent that the slides were not all that was said at the training sessions for CRAs or sites, and I do not consider that it has been shown that what was said about IC6 was inadequate. There do not appear to have been complaints about the inadequacy of these sessions.
361. Insofar as the criticism is that IC6 was not broken down in the training materials, I consider it to be answered by the fact that in the Data Collection Worksheet IC6 was broken down. The Data Collection Worksheet was prepared with input from Cardiorentis, and was made available to all sites. Not all the sites used it, but it was available to all either to use or to produce an equivalent, had they wished to do so. Professor Zannad fairly described the breakdown of IC6 in the Data Collection Worksheet as ‘well done’, and said that it ‘does not seem that there was anything wrong with the Data Collection Worksheet’.
362. The second strand was that the identification of IC6 deviations by IQVIA’s monitoring was late and incomplete, and so, as a result, was the information supplied to Cardiorentis. This itself had two aspects.
363. The first aspect was that monitoring visits were late, and deviations, even if detected by the CRA, were not always so detected on the first visit. I have already considered this part of Cardiorentis’s case. Cardiorentis did not plead a case relating to particular failings of specific CRAs to carry out monitoring visits in a timely or appropriate manner. Without such an examination I do not consider that it is possible to conclude that monitoring visits should have been conducted sooner or differently with the result that IC6 violations would have been identified more quickly or more completely than they were. I have already set out that there may have been various reasons why a visit was delayed, or monitoring could not have been effective on a particular occasion, for reasons which were not the fault of the CRA or IQVIA more generally.
364. Furthermore it appears clear that identifying an IC6 deviation from a review of records such as the CRAs were performing is in many cases a difficult one. This is suggested by the number of such deviations which were not identified, at a wide variety of sites in a large number of countries, by different CRAs. It is also suggested by a consideration of the nature of the exercise involved, which involved some elements of medical judgment. These included: (i) if a patient had received an IV bolus less than two hours prior to randomisation, it would be necessary to know whether it was a diuretic, which might have been obvious, but might not have been; and (ii) at least after the adoption of Protocol version 03, if there had been a change in IV medication during the two hours prior to randomisation, whether the changed medication was being administered to treat HF.
365. I consider that it is also suggested by the fact that a significant number of IC6 deviations were not identified by the various processes of co-monitoring, remote reviewing and auditing which Cardiorentis or its contractors performed, or during the inspections which certain regulators carried out. I treat this point with caution. I recognise that the work involved in these processes was dwarfed by that which IQVIA did and was required to do; that they were not designed to take the place of IQVIA’s monitoring; and that their overall objectives were at a higher level, to obtain a degree of assurance of compliance. Nevertheless, eligibility deviations were one of the matters being considered. In relation to the remote review, the Work Instruction stated that its focus would include protocol deviations, including signs of increased AHF therapy. In those circumstances the number of cases of violations of IC6 identified in the tables which IQVIA produced which had not been identified by these processes did indicate that such identification was by no means straightforward, and counted against any inference that a failure to identify an IC6 deviation must have been the result of negligence.
366. The second aspect of this strand of Cardiorentis’s argument is that there was a delay in violations identified by CRAs being reported in PD Logs, including by reason of a gap in the production of such logs between May and October 2013. There may have been breaches of contract by IQVIA by reason of these matters, but it is difficult to attach causative significance to them. There is no reason to consider that had they been reported earlier in PD Logs IQVIA would have acted differently from how it did. As to Cardiorentis, when the deviations were reported in the PD Logs, it was apparent what that deviation was and when the randomisation had occurred. Cardiorentis could have responded to both. In fact there is no evidence that it complained about delays in such deviations being reported to it, and no suggestion at the time that it would have done something different had such deviations been reported in PD Logs earlier than they were.
367. The third strand was that particular attention needed to be paid by IQVIA to ensuring effective monitoring of IC6 by reason of various matters which occurred during the course of the study. I have already identified the principal matters in my review of the facts relating to IC6.
368. I do not consider that the mention in the Project Risk Log of the ‘study design’ risk referred to above is of particular relevance. The specific concern addressed was a risk that the final evaluation as to whether the patient was still symptomatic might be overlooked by sites. The answer to this concern given in the Risk Log was ‘Training’, and this appears appropriate. In any event, this issue does not appear to have been a particular problem during the course of the trial.
369. The second matter was the identification by CRAs of IC6 deviations, including at an early stage in the study. Cardiorentis contended that, upon such identification, a root cause analysis ought to have been carried out. It pointed to the slides prepared by Veronica Sandstrom as to ‘TRUE-AHF Monitor’s Responsibilities’, which included that CRAs were expected to carry out an analysis when encountering problems to identify the root cause, the significance of the problem, and the corrective and preventative action, and should report on the matter.
370. It is clear, however, that what would be necessary by way of root cause analysis, and a response to a root cause analysis, would depend on what the problem was which presented itself to the CRA. In the present case I was shown what was done by Ms Landolfi in relation to Rome site 1605: her site visit report shows that she identified the cause of a deviation, its severity, took action by speaking to the Investigators and retraining site staff; concluded that no further action was still required; and noted that the issue had been reported to the IRB/IEC. I do not consider that that analysis can be said to have been deficient; and indeed it was not suggested to be so by Cardiorentis. Yet that sort of analysis might have been done in many cases: no evidence was adduced as to what had occurred in the vast majority of instances when IC6 deviations were identified. I do not consider that there is a basis for concluding that CRAs failed to conduct adequate analyses of the cause of IC6 deviations which they encountered.
371. A further matter relied on by Cardiorentis was the mention of IC6 in the Remote Monitoring Issues Log, dated 14 October 2013. If the ‘Instructions’ for the use of the Issues Log were complied with, that entry, coupled with the Response annotation ‘Y’ showing that this item had been closed, tends to indicate that the issue had been considered with the relevant CPM, and that the issue was then discussed in a weekly quality management call. Assuming that to have been the case, then there was a discussion between the parties as to how to address misunderstandings of IC6. In the circumstances, and although the picture as to what happened is not complete, I do not consider that it can be concluded that IQVIA was at fault in failing to take any further steps in response to IMR’s raising of the issue at that stage.
372. The final particular matter relied upon in Cardiorentis’s Closing Submissions was that, as a result of the reporting of IC6 deviations in Site 1605, it was, by December 2013, known to two of IQVIA’s senior Medical Advisers that sites were experiencing uncertainties as to the application of IC6, and that this should have been dealt with, including by clarification in the CRA newsletters.
373. The communications arising from the identification of IC6 deviations in Site 1605, and in Polish Site 1807, were not pleaded, nor were they referred to in the witness statements, even though Dr Holzmeister was involved in giving advice in relation to the issue which had arisen as to the last part of IC6. It appeared to me that the full picture had not been put before the court. It seems at least possible that it was in part as a result of the raising of the issue of the ambit of medications referred to in the last sentence of IC6 that there was an amendment of its terms in Protocol version 03. That was not, however, explored. What does appear clear is that Cardiorentis, as Sponsor, and Dr Holzmeister in particular, was made aware of an issue as to whether the last sentence of IC6 covered infusion of all medications or was limited. I do not consider that it is safe to draw any conclusion from this unpleaded and uninvestigated area that IQVIA was at fault in not taking further steps to clarify to CRAs the application of the last sentence of IC6. Cardiorentis has made no clear case as to what ‘clarification’ would have been appropriate as a result of what was learned in relation to the Rome and Polish sites; and it is in any event speculative as to whether any such ‘clarification’ would have had an effect in preventing IC6 deviations.
Non-EC3 or IC6 eligibility deviations
374. Some patients were randomised with eligibility deviations other than breaches of EC3 or IC6. In the SCI analysis, 64 patients were treated as having breached eligibility criteria other than EC3 or IC6. In Ms Harper’s Exhibit 6, 66 patients were treated as being in this category. In the Agreed Schedule 60 patients are said to have ‘other’ eligibility deviations, though some 12 of those also had EC3 or IC6 deviations. The instances shown in the Agreed Schedule of ‘other’ eligibility deviations occurred at 37 sites in 17 countries.
375. Cardiorentis pleaded no specific case in relation to eligibility deviations other than EC3 or IC6. Ms Harper in her reports did not deal in any detail with these other deviations, and accepted in her evidence that she had not looked closely at them. In essence, therefore, Cardiorentis’s case in relation to these other deviations was confined to its general case as to failures of training and monitoring which I have already considered above, and which I have found did not constitute a basis for concluding that there were breaches on the part of IQVIA which caused ineligible subjects to be enrolled.
376. Looking specifically at the category of patients who were enrolled with eligibility deviations other than EC3 or IC6, there are particular reasons for rejecting a case that these must have occurred by reason of breaches on the part of IQVIA.
377. In the first place, they were diffuse in nature. According to Ms Harper’s Exhibit 6, they involved breaches of Exclusion Criteria 1, 5, 7, 9, 10, 11, 12 and 17; of Inclusion Criteria 1, 2, 3 and 5; and 3 other forms of deviations in eligibility criteria. The largest number shown in that table is in relation to breaches of Inclusion Criterion 2, which is set out above. As is apparent from its terms, Inclusion Criterion 2 was itself composite, in the sense that it required three different metrics to be established in order for a patient to be classified as suffering from ADHF, and could have been breached in various different respects.
378. They were also diffuse as to where and when they occurred. They occurred in small numbers at a considerable number of sites, and fairly steadily over the period of the study.
379. These two points suggest that they were the sort of mistakes which Investigators will make from time to time when conducting a study of this type, in the pressured environment of an emergency setting. It is also difficult to accept that they point to any specific defect in the training of sites which was provided, for almost all sites randomised many patients who were not ineligible for any of these reasons.
380. Secondly, most of these deviations were in fact identified by CRAs, and included in CTMS and the PD Logs. Only 4 of the 66 cases in Ms Harper’s Exhibit 6 were not identified in this way. This was therefore not a case in which it could be said that the process of monitoring by CRAs had failed to detect these deviations at all.
381. Cardiorentis sought to make the case, nevertheless, that the identification of these deviations was too slow. As part of its Closing Submissions, it produced a Schedule which set out what it contended were delays in the identification of these eligibility deviations, demonstrating that monitoring was ‘endemically too slow’. Mr Kitchener QC objected to Cardiorentis seeking to rely on the contents of that Schedule or the case made by reference to it, given that it was adduced only after the close of the evidence. I considered that, at least in relation to sites which were not mentioned at all in Cardiorentis’s pleading, that objection was valid. To the extent that a case was being made that there were specific delays in identification of such deviations which were the fault of IQVIA, that case would have needed to be articulated at a time when IQVIA could have dealt with it. Furthermore, and in any event, the Schedule did not appear to indicate that, in any significant number of cases, earlier identification would have been likely to have prevented other deviations. Largely, as I have said, this category of deviations did not involve repeated failings by sites.
382. One specific case of two similar eligibility deviations at the same site was explored to some extent in the evidence. This was in relation to patients 1205004 and 1205019, at site 1205 at Bad Nauheim in Germany. In each case the patient was randomised although their creatinine clearance was slightly below 30 at screening and there was therefore a deviation in relation to EC5. Patient 1205004 had been randomised on 24 May 2013. According to a Report of a Site Visit which took place on 6-7 August 2014, this deviation was first noted by a CRA on 13 August 2013, although it first appears in a PD Log on 4 September 2014. The Site Visit Report records that the issue was discussed with the Investigators who were reminded not to enrol patients who did not meet the criteria. Patient 1205019 was enrolled on 6 January 2014, and the date on which the deviation was noted by the CRA is recorded as 3 June 2014. There is a similar note as to a discussion with the Investigators.
383. If the deviation in relation to Patient 1205004 was identified in August 2013 and the discussion with the Investigators in relation to that patient occurred shortly thereafter, then the second patient would have been enrolled notwithstanding that discussion. It is impossible to conclude that such enrolment would have been the fault of the CRA. If on the other hand, as Cardiorentis at one point suggested, the deviation in relation to Patient 1205004 was only identified in August 2014, then this might have been a case where another patient was randomised with the same eligibility deviation between the occurrence of the original deviation and its identification on monitoring. It is not possible, however, to say that this is more likely than not to have been the case; and in any event it is not possible to say that the CRA was at fault in not identifying this issue earlier; and I regard it as speculative whether, even if the issue had been identified in relation to Patient 1205004 earlier it would have prevented the eligibility deviation in the second patient.
384. Thirdly, the only real ‘cluster’ of eligibility deviations in this category was those of site 1605 in Italy. Five patients with non-EC3 or IC6 deviations were randomised between 11 and 22 December 2012. An additional four patients with such deviations were randomised between 5 January and 11 February 2013. All these deviations were identified by the CRA on 11-12 February 2013; and a final deviation, in a patient randomised on 11 February 2013, was identified on 18 February 2013.
385. I do not see how these eligibility deviations can be credibly attributed to fault on the part of IQVIA. It is difficult to see that they were caused by a failure in the training provided, given that similar deviations were not found in significant numbers in other sites, given also what is recorded as to the nature of the training of the site, and that the Principal Investigator at this site, Professor Di Somma, was a member of the Steering Committee and should have had a good knowledge of the Protocol in any event. Equally, the monitoring does not appear to be at fault. The deviations were identified promptly after occurrence.
386. I have already set out my conclusions as to the contractual responsibility on IQVIA as to source data verification.
387. In the present context, Cardiorentis’s case as to inadequate or belated SDV adds little to its case on inadequate or belated monitoring. SDV was a part of the monitor’s responsibilities, and an integral part of the monitoring activities. What I have said above in relation to Cardiorentis’s general case on monitoring applies to its case on late and inadequate SDV.
Alleged breach of CQA
388. Cardiorentis’s case is that IQVIA was in breach of clauses 4.4.1 and 4.4.2 of the CQA in not processing quality issues in accordance with operating procedures and in not notifying quality issues to Cardiorentis timeously.
389. Insofar as the case here was that IQVIA had not processed issues properly, this was, as I understood it, essentially a complaint that eligibility deviations had not been properly and promptly identified and notified to Cardiorentis. As such this case did not add anything significant to Cardiorentis’s case under the GSA.
390. Furthermore, I consider that clauses 4.4.1 and 4.4.2 of the CQA are concerned with the reporting of known quality issues. As set out in its clause 1, the purpose of the CQA was to facilitate the sharing of information between Cardiorentis and IQVIA. Its purpose was not to impose investigation obligations. Insofar as eligibility deviations had been identified by CRAs, they were reported in the PD logs, and were known to Cardiorentis. Insofar as they were only identified by the BIOS searches which were performed as part of the BDR, any quality issue which they constituted was not one which could have been reported earlier because it was not known to IQVIA.
391. In relation to clause 5.2 Cardiorentis’s pleaded case was that QA to QA meetings or teleconferences had not been on a monthly basis, and had not dealt with quality issues, audits or project level questions adequately. Clause 5.2 does not, however, impose an obligation to deal with quality issues, audits and audit CAPAS or project level questions ‘adequately’. It is concerned simply to stipulate that there should be monthly QA to QA meetings or teleconferences at which these matters would be discussed. An obligation on IQVIA (alone) to deal with matters ‘adequately’ cannot be read into this. Insofar as Cardiorentis’s complaint was that meetings/teleconferences had not occurred every month, that was a matter of which Cardiorentis was aware and presumably it could have scheduled meetings had they been thought necessary. No case was made out that any identifiable eligibility deviation(s) had been caused by an absence of monthly meetings.
Other allegations of defective performance
392. Cardiorentis pleaded that there had been ‘irregularities concerning the Kansas Medical Center’, and that as a result of IQVIA’s breach of contract, it did not have a reliable understanding of these.
393. The irregularities concerned were revealed as a result of an FDA inspection of the site in June and July 2016. The irregularities which it was pleaded that the FDA had identified were that in relation to two patients who had been randomised into the trial ECGs sent into the central reader were not those of the patients concerned (or of any patient in the study). In both cases non-study patients’ ECGs had been redacted and relabelled with a study patient number; in one case that was not corrected; and in the other the correct ECGs were ultimately submitted.
394. I was unable to conclude that these matters, which may have involved an element of fraud on the part of the site, were ones which the CRA would have been able to, or should, have identified at the time. This was essentially for the reasons given by Ms Queiroz in her evidence. In any event, it did not appear to have any relevant causative significance. The agreed schedule of Patients Randomized by Site indicates that no patients with eligibility deviations were randomised at this site after the enrolment of the second of the two patients to which this issue related.
395. Insofar as other features of what happened at Kansas site 0401 were used by Cardiorentis in its Opening as the basis for a more general argument that the way in which the study was conducted was one in which ‘chaos and confusion and muddle abound’, I considered that this went beyond the pleadings. I should however record that the evidence which I heard did not lead me to the conclusion that, overall, there was abounding ‘chaos and confusion and muddle’ in the way in which the study was conducted by IQVIA. Furthermore it was apparent that many of those involved in the study were extremely thorough and painstaking: Ms Raskino and Ms Queiroz may be taken as examples.
396. My conclusion is that Cardiorentis has not succeeded in showing that any identifiable or significant number of eligibility (or other) Protocol deviations were the result of IQVIA’s breach of contract. While there were some limited areas in which breach was established, as I have set out, most of Cardiorentis’s allegations of breaches which caused there to have been the enrolment of subjects with eligibility or other protocol deviations were not made out.
The BDR Process
397. It is convenient next to consider Cardiorentis’s case in relation to the conduct of the BDR. It was not a part of this case that deficiencies in this process had led to the inclusion of ineligible subjects in the study: clearly it did not, given that it was conducted after the last patient had been enrolled. It is equally not the case that, had it been performed differently, that would have led to Cardiorentis being able to take any significant steps in relation to the conduct of the study. By the time that the database was locked, and indeed for some time prior to that, there was little that Cardiorentis could have done, even had it sought to do something, about the number of eligibility or other protocol deviations.
398. Realistically, in his closing submissions, Mr Stanley QC said that Cardiorentis had no contractual complaint about the way in which the BDR process was handled. But Cardiorentis maintained a case that the BDR had not been properly performed and this meant that the per protocol population cannot be taken as a reliable indication of the effect of ularitide on a population without eligibility deviations or as a useful cross-check on the results on the ITT population.
399. This case was that, as it was actually carried out, the BDR process was messy, muddled, confused and botched. It is necessary to evaluate this case with some care.
400. I have already set out the basic facts in relation to the BDR process. There can be no doubt that the course it followed was not that recommended for a BDR process in the paradigm set out in Ms Leipoldt’s slides of 13 November 2013. It was less organised and methodical and less well documented. Pressure of time contributed to this.
401. There remains the question as to whether this meant that the results of the BDR process were flawed, in the sense that the results of the per protocol set it produced were not such as to provide enhanced confidence in the results of the ITT set.
402. As already set out, one decision made during the BDR process was that the period of 2 hours referred to in the last sentence of IC6 should be taken as two hours prior to infusion, rather than randomisation. The result of this was that only patients who had received an IV bolus of a diuretic, or a change in infusion rates of medication to treat HF within two hours prior to the start of infusion would be excluded from the PP population. As Cardiorentis pointed out, the effect of this was both to mean that some patients who had eligibility deviations in accordance with the Protocol were included in the PP set and also that some patients were excluded from the PP population, even though they had not had eligibility deviations in accordance with the Protocol.
403. The justification for this decision, as set out in the BDR Plan, was that the intention of the relevant part of IC6 was to make sure that the patient had not received, within 2 hours of receiving the study drug, any medication that could confound the effects of the study treatment. The Protocol had used t0 as the start point for observations in relation to Endpoint 1. Slides presented by Dr Packer, including at the Heart Association in Paris in April 2017 confirm that the intent of IC6 was to judge eligibility by reference to t0. Accordingly, the decision appears justifiable as tending to produce a PP population which matched the scientific model of the study. This was the evidence of Dr Sager. It was a decision made, I am in no doubt, in good faith, by both IQVIA and Cardiorentis representatives on the BDR teams, who took it blind as to whether patients had received ularitide or placebo. Though it can be said not to be in accordance with the precise words of the Protocol and SAP, I do not consider that it detracted from the usefulness of the PP set.
404. A second decision was to use only BIOS listings to identify EC3 deviations, which meant that some 23 patients identified in CTMS as contravening EC3 were not included in the PD Spreadsheet and not excluded from the PP population. The decision to use only BIOS listings for this purpose may have been on pragmatic grounds. As the omissions were effected ‘blind’ to whether the patients concerned had taken ularitide or placebo, it should not have significantly undermined the reliability of the PP set.
405. A third matter was the fact that 10 patients identified in the BIOS listing for patients who had had prohibited medications 7 days prior to t0 had not appeared in the PD spreadsheet. This appears to have been an error, as Ms Raskino acknowledged. It was not relied upon by Cardiorentis as a breach of IQVIA’s duty, and did not in fact make a difference to the composition of the PP set because these 10 patients were excluded from the PP set for other reasons.
406. A fourth matter was what was termed during the hearing ‘the furosemide issue’. This was a reference to the fact that BIOS programs had revealed that in a considerable number of cases there was no entry made by the site on the Previous and Concomitant Medications (CONMED) page of the CRF that furosemide had been administered after the start of emergency services. In these cases, nevertheless, the site will have marked IC6 on the IE page as being satisfied, which itself referred to the patient having received IV furosemide or equivalent diuretic at ≥40 mg after the start of emergency services.
407. The decision made during the BDR process was that such BIOS listings should not be used for determining whether a patient had or had not satisfied IC6 in this regard, and that there should be exclusion from the PP set only if there was a reference in CTMS to a failure to satisfy the criterion. I have already set out the way in which this was explained in the BDR Plan. The decision made was that the lack of an entry for furosemide on the CONMED page was likely to be a failure of data entry, not of actual non-compliance with IC6. That appeared to me to be an understandable and reasonable decision. The evidence established that furosemide (or equivalent) is given to most patients with ADHF. In Professor Zannad’s first report it is said that ‘Using furosemide is an extremely effective and rapid way of relieving shortness of breath and fluid retention.’ It is likely that most patients presenting with persisting dyspnoea at rest will have been given furosemide after the start of emergency services. That the issue was in essence one of data entry rather than non-compliance with IC6 was indicated by the results of Cardiorentis’s remote review exercise. The final report indicated that of 58 patients who had been identified as having potential issues as to compliance with IC6, 52 had been resolved, and only 2 had been found to have deviated in relation to the first part of IC6.
408. Insofar as Cardiorentis sought to widen the furosemide issue into a complaint that the point as to non-completion of CONMED pages to record furosemide had not been brought to its attention at an early enough stage, or that it indicated that SDV was not being properly performed, IQVIA contended that this was a matter which had not been pleaded, and which Cardiorentis was seeking to raise in an unfair manner. I considered that that objection was correct.
409. A further aspect, overlapping some of the previous matters, was that Cardiorentis suggested that the decisions made in the BDR process may, at least subconsciously, have been affected by a bias in favour of keeping patients within the PP set. I do not consider that this is made out. The process of seeking BIOS listings was an exercise which was very likely to (and in fact did) uncover additional Protocol deviations. Exclusions from the PP population were made of some patients who complied with the letter but not the spirit of the Protocol, as in the case of patients who had received an IV bolus of a diuretic or whose infusions had changed in the two hours prior to infusion but not prior to randomisation. In other respects as well, as Dr Sager said, decisions on the BDR were conservative, in the sense of omitting patients from the PP population who might have been included. An example was the decision to exclude from the PP population all patients who had received a PDE inhibitor within 72 hours after randomisation irrespective of the reason for its administration. Further it was Ms Raskino’s evidence, which I accept, that those involved in the process were not seeking to minimise the number of patients excluded from the PP population, but to produce a set which excluded those with clinically relevant protocol deviations.
410. My overall assessment of the BDR process is that, while not perfect, it was one which was conscientiously undertaken by those involved from both Cardiorentis and IQVIA. It represented a professional attempt to identify a set of patients which excluded those who had clinically relevant features which meant that their inclusion was not in accordance with the scientific model on which the study was built. It had, importantly, the significant advantage over post hoc analyses of being conducted blind as to treatment status.
Was the study unreliable or of little use by reason of inclusion of patients with eligibility or other protocol deviations?
411. I have concluded that Cardiorentis has not established that any identifiable or significant number of eligibility or other protocol violations were caused by breach of contract on the part of IQVIA. I will go on in this section to consider the position if I am wrong in relation to that conclusion. If there were eligibility or other protocol violations which were caused by breach of contract, what were the consequences of that? This question is complicated by the fact that there can be no dispute:
(a) That Cardiorentis had not contracted for the conduct of a study which produced a particular result. It had contracted instead for the conduct of a study which produced a meaningful and interpretable result, positive or negative, as far as the efficacy of the drug was concerned;
(b) That some eligibility and other protocol deviations were to be expected in any study; and
(c) There was, as I have already found, no recognised threshold or cut off in the number or percentage of eligibility or other protocol deviations which would mean that regulators or the scientific community would consider the study to be unreliable.
412. In those circumstances, IQVIA argued that Cardiorentis could not show that, even if there were eligibility or other protocol deviations which were its fault, they had any adverse effect. The study was still a robust and interpretable study, albeit one which was negative as far as the efficacy of the drug was concerned. Even had there been no breach and no resulting eligibility or other protocol violations, Cardiorentis would have had robust, negative, results.
413. I have concluded that IQVIA is clearly correct in relation to this aspect of the case. My reasons for this follow, under eight overlapping heads.
414. In the first place, there can be, and was, no serious dispute that the study was, reliably, negative in relation to Endpoint 2 (cardiovascular mortality). The SCI Report itself stated that the study had produced a reliable database. The NEJM article concluded without caveat that ularitide failed to protect against long term myocardial death. Dr Packer wrote an article in the NEJM for August 2019, in which, in this context, TRUE-AHF (and RELAX 2) were said to be ‘definitive’ studies. Dr Sager’s evidence was that the study was ‘undoubtedly negative in relation to endpoint 2’. Professor Zannad accepted that the negative result was reliable, giving evidence that in relation to long term mortality ‘it is unlikely that short-term administration would be beneficial, at least in these conditions of this trial with this kind of population’ (Day 17/26).
415. The inclusion of ineligible patients did not affect this negative result. As the results were set out in the NEJM article, in the Full Analysis Set there were 236 (21.7%) cardiovascular deaths in the ularitide group and 225 (21%) in the placebo group; with a p-value indicating the degree of likelihood of the result being the product of chance of 0.75. The results set out in Supplementary Table S4 for cardiovascular mortality, excluding ineligible patients, were 186 (20.8%) in the ularitide group and 187 (20.7%) in the placebo group; with a p-value of 0.87. Thus, the inclusion of ineligible patients did not lead to any material difference to the negative result in relation to Endpoint 2.
416. The results in relation to mortality Endpoint 2 were consistent with the absence of any significant difference between the ularitide and placebo groups as to changes in cardiac troponin T levels during infusion. As Professor Zannad accepted, the results in relation to troponin levels were not affected by the presence of ineligible patients in the study. Taken together, these matters were soundly-based evidence suggesting that the hypothesis that the administration of ularitide reduced myocardial injury by attenuating cardiac wall stress was incorrect.
417. IQVIA submitted that these matters had a further significance. It contended that the same myocardial injury hypothesis was being tested in relation to Endpoint 1 as in relation to Endpoint 2. It contended further that the results in relation to Endpoint 2, and in relation to cardiac troponin T levels, effectively answered the sole scientific theory on which the testing of Endpoint 1 was based. IQVIA pointed, amongst other things, to Supplementary Figure S1 to the NEJM article, to some of Dr Packer’s responses to the NEJM peer reviewers, and to passages in Professor Zannad’s reports, to support the argument that the falsification of the hypothesis as to myocardial injury gave a reliable negative answer to the question being asked by examining Endpoint 1.
418. I was left in doubt as to whether the position was quite as simple as that. It was an area in which I considered that assistance would have been obtained by sight of the entirety of the correspondence with the FDA and also had Dr Packer given evidence. I accept that the myocardial injury hypothesis was a part of why Endpoint 1 was being looked at. Yet it appeared to me that Endpoint 1 was also being looked at simply to see whether ularitide provided symptomatic relief (over and above that provided by standard treatment), and reduced the short term occurrence of adverse events, whatever the mechanism for that might have been. That was suggested by certain comments by the NEJM peer reviewers. I also understood this to be Professor Zannad’s evidence when cross-examined.
419. I consider that it is sufficient for the purposes of the issue I am addressing to say that, if IQVIA were correct that the results in relation to Endpoint 2 gave a reliable answer to the sole question being posed by testing Endpoint 1, then this would add to the force of this first point.
420. Second, the effect size for Endpoint 1 which the study was powered to analyse was 12.5%. Dr Holzmeister himself gave evidence that that was a figure which had been discussed with the FDA as ‘a meaningful threshold to get the drug approved’; was not an arbitrary number; and was ‘a definitive number’ so that, if not met, in 99% of cases the drug would not be approved (Day 3/124). (I did not accept that that was in answer to questions which Dr Holzmeister could not have expected, given the prominence attached to effect size in IQVIA’s experts’ reports and Opening Submissions, and given that the court had indicated that the derivation of the figure of 12.5% was a matter of interest at Day 2/8-9).
421. The results of the study, as published in the NEJM, showed, in the Full Analysis Set, an effect size, namely the percentage of the population which had improved in respect of Primary Endpoint 1, of 0.9%. Omitting ineligible patients, the results, as set out in Supplementary Table S4, is of an effect size of only about 1.9-4.0% (Sager, first report paragraph 118).
422. I accept the evidence which Dr Sager gave that a demonstration of an effect size of, or at least of the order of, 12.5% would have been of great importance to the approval of the drug, in particular for two reasons he gave: (1) that a few percent improvement in symptoms that are transient is minor and would not have been seen as a significant benefit, especially (2) when what is important for approval is the benefit to risk relationship, and where there was good evidence of an increase of hypotension and drug discontinuation in patients receiving ularitide by comparison with placebo. Thus, given the known existence of hypotensive effects of the drug, a positive effect in relation to Endpoint 1 had to be of significant size to make the drug likely to be approved.
423. Because the study was so overpowered in relation to Endpoint 1, the ineligible patients can, for the purpose of analysis, be removed, without the study falling near to (and certainly not below) the number of evaluable patients necessary to test for Endpoint 1. It is clear that the results, even omitting all ineligible patients, indicated an effect size which was far from 12.5%. It was not shown that there was any set of realistic or plausible assumptions as to what might have been the effect of substituting ineligible by eligible patients which would have yielded an effect size of 12.5% or in that region.
424. In those circumstances, even if it is assumed that all the ineligible patients were randomised as a result of IQVIA’s breach, there is no ground for thinking that the inclusion of those patients (or the non-inclusion of eligible patients who might have been recruited in their stead) meant that the study failed to show an effect size of the order contemplated by the Protocol and discussed with the FDA. Instead, the study provided reliable evidence that there was not an effect size of that order.
425. Thirdly, even for the small effect size in relation to Endpoint 1, the study failed to meet the pre-specified level of statistical significance of 0.01%. This is so, even on the analyses which exclude all the ineligible patients who were identified and excluded by SCI. Such exclusion of all ineligible patients is an unscientific exercise, given that there is no biological plausibility in treating all eligibility deviations as a single group and all patients with other protocol deviations, or none, as another. Nevertheless, if that exercise is done, the nominal p-value, as shown in Table S4 of the Supplementary Appendix to the NEJM article, is 0.035. That result is based on an analysis without the multiple imputations for missing data specified in the SAP. With multiple imputation the nominal p-value would be 0.074.
426. Accordingly, even omitting all ineligible patients, the study would, as I have said, have failed to show a result which met the specified level of statistical significance. I accept Dr Sager’s evidence that a trial such as the present would be regarded as positive only if the relevant endpoint had been achieved to the prespecified level of statistical significance, and would be regarded as negative if it did not; and that there would be no proper basis for disregarding the pre-specified p-value (Sager, Supplemental Report, paras, 19(b), 23). On that basis, even omitting all ineligible patients the study would have produced a negative trial.
427. It is also germane to consider what would be the position if I was wrong as to only some of my findings in relation to whether fault on the part of IQVIA had led to the randomisation of ineligible patients. What would be the position if, for example, it were the case that it was as a result of IQVIA’s fault that there were the EC3 violations, but not the others; or the IC6 violations but not the others; or EC3 and IC6 violations but not the others? The position, as set out in the SCI analysis, is that omitting patients with EC3 violations shows an effect size of about 2%, with a nominal p-value of 0.21; omitting patients with IC6 violations shows an effect size of 1.9% with a nominal p-value of 0.16; omitting patients with EC3 or IC6 violations showed an effect size of 2.8% with a nominal p-value of 0.10; and omitting all ineligible patients showed an effect size of 4% with a nominal p-value of 0.035.
428. This illustrates that it is the category of deviations which were not EC3 or IC6 violations which were, in Mr Kitchener QC’s phrase, doing a lot of the ‘heavy lifting’ in relation to reducing the nominal p-value to one which approached statistical significance. Given that Cardiorentis’s case in relation to IQVIA’s responsibility for non-EC3 or IC6 deviations was, in my judgment, and for the reasons I have given, particularly weak, this is an important indicator that the inclusion of patients whose ineligibility could be blamed on IQVIA did not produce the study’s negative results.
429. Fourth, in my view the best way available to the court of checking whether the randomisation of subjects with protocol (including eligibility) violations may have had a material effect on the results of the trial is to look at the results of the Blind Data Review process, and the per protocol population which it produced. As I have set out above, it was a pre-specified part of the Protocol and the SAP that there should be a Blind Data Review which would consider which protocol deviations were sufficiently significant to merit their exclusion from the per protocol population, and it was agreed between Dr Sager and Professor Zannad that such a review is a standard and important part of a clinical trial. It was a process carried out before unblinding, and therefore was not biased by knowledge of whether the patients included or excluded had received ularitide or the placebo.
430. It is apparent simply from reviewing them, and considering how they could occur, that not all protocol, and not all eligibility, deviations will have any clinical significance. Dr Sager and Ms Harper agreed that there was no blanket rule that every patient who does not meet any eligibility criterion represents a major protocol deviation. A Blind Data Review is aimed at producing, as objectively as possible, a decision as to whether a protocol deviation is likely to be of any significance to what the trial is looking at, and to exclude from the per protocol population only those where the deviation was of such a kind.
431. As set out above, the results of the study on the per protocol population, which involved the exclusion of 395 patients from the ITT population, were of an effect size for Endpoint 1 of approximately 2.8% with a p-value of 0.137. 159 of the 358 patients with eligibility deviations considered by SCI were included in the per protocol population. Excluding only those 199 patients who had eligibility deviations which were regarded in the BDR as meriting exclusion from the per protocol population, yields results of an effect size of 2.8% with a nominal p-value of 0.215. This is a long way from the effect size contemplated, or the p-value pre specified, in the Protocol and SAP.
432. Fifth, there was not shown to be any biologically plausible explanation as to why the patients who were ineligible should have responded differently to ularitide (or placebo) from eligible patients. Cardiorentis pleaded no such case in relation to the patients who had eligibility deviations other than IC6 or EC3. In relation to IC6 deviations, Cardiorentis had pleaded that patients who breached IC6 may have been over-exposed to diuretics, but this was not a case supported by evidence from Professor Zannad, and was not put to Dr Sager. No other case in relation to there being a biologically plausible explanation for a difference in outcomes between subjects randomised with IC6 deviations and those without was supported by expert evidence. There is, furthermore, no reason to believe that they will have shared relevant characteristics with patients who breached EC3.
433. In relation to patients with EC3 deviations, two theories were put forward. One was that the use of aminophylline and theophylline, which are bronchodilators, may have confounded the assessment of dyspnoea. In his oral evidence Professor Zannad said that he did not consider that this was likely to be important. Dr Sager gave cogent reasons as to why it was an unconvincing theory, in that aminophylline/theophylline relieves the bronchospasm that can accompany ADHF and which can continue even when ADHF has subsided. The effect of treatment with aminophylline/theophylline would not, of itself, mean that patients would count as markedly or moderately improved, unless there had been an improvement in the underlying HF; while on the other hand, treatment with aminophylline/theophylline, by relieving bronchospasm might, if anything, make a positive effect from ularitide more apparent.
434. The other theory put forward was that a patient receiving aminophylline/theophylline, with ularitide, would have been more likely to have suffered from hypotension. I did not consider that this was a plausible explanation for any difference of effect. If a patient suffered from hypotension after infusion (s)he would be likely to appear as ‘worsened’. Only 2 patients in the EC3 group who had received ularitide, rather than placebo, were classified as ‘worsened’, which was a smaller number, and percentage, than amongst those having taken the placebo. If, on the other hand, the receipt by the patient of the prohibited medication, with ularitide, had an effect which did not impinge on the clinical composite, then it would not have impacted the results for Endpoint 1. The evidence of Dr Sager, which I accept, was that there was, in fact, no significant difference between the proportion of ularitide patients who experienced hypotensive adverse events among eligible patients and ineligible patients.
435. Sixth, the results of the trial were consistent with the results of ASCEND-HF in relation to nesiritide, and in particular the fact that only a small effect size, which Dr Sager described as ‘clinically unimportant for a transient symptom’, and which did not achieve the prespecified level of statistical significance, was detected in relation to dyspnoea at 6 or 24 hours. They were also consistent with the results of GALACTIC, which indicated no significant difference between study therapy (intensive treatment with nitrates and other approved vasodilators) and placebo groups in relation to dyspnoea at 2 or 6 days; and also with the results of RELAX 2 in relation to serelaxin and mortality.
436. In this regard I accept the evidence of Dr Sager that the primary effects of ularitide, nesiritide, serelaxin and nitrates is vasodilation; that these drugs all have similar haemodynamic effects; and thus that the evidence from the four studies is strong that the effects of vasodilation, of acute unloading of the heart, decreases in wall stress and decompression of the heart, do not have meaningful short or long term benefits in treatment of ADHF (Joint Memorandum of Cardiology Experts, paras. 11-12).
437. Seventh, there has been a general acceptance in the scientific community, as a result of the studies, including TRUE-AHF, ASCEND-HF and GALACTIC, (and also RELAX 2 in relation to mortality) that drugs like ularitide do not have a significant effect over and above background therapy in reducing symptoms, CHF worsening or mortality. Dr Sager put the matter in this way in his first report.
‘[82] Although the trials clearly demonstrated that acute vasodilatory therapy in ADHF does not reduce symptoms, CHF worsening, or mortality, these trials are very important (and were very valuable) in that the information permits cardiologists and drug and device developers to focus on more health beneficial approaches, such as therapeutic modalities to prevent ADHF episodes from initially occurring.
[83] Given that none of these vasodilator therapies showed a clinical benefit, the consensus view of the cardiology scientific community is that acute vasodilator therapy is a futile area for further investigation as a treatment for ADHF. To the best of my knowledge, no company is still conducting trials of vasodilators in relation to ADHF.’
438. I accept this evidence of Dr Sager, which was not contradicted by any published material.
439. Eighth, the evidence of the expert cardiologist called by Cardiorentis, Professor Zannad, was of significance. Professor Zannad said in his first report that, at the time the results of TRUE-AHF were uncovered, there was still a hope that ularitide and similar drugs would be effective treatments for ADHF, but that ‘now, in 2021, there have been other trials in this space that have been unsuccessful’ (para. 9.6.2).
440. His oral evidence was that he, in common with many in the cardiological community, had concluded that drugs like ularitide were not going to be seen to be efficacious if tested on the type of population involved in the TRUE-AHF study for the endpoints specified in that study, though it might be shown to have an effect if tested on a different population and/or for different endpoints and/or in a different way (Day 17/42, 44, 76-78). The reason why ularitide had not shown an effect in the study was, he considered, ‘related mainly to the product in itself’ (Day 17/146).
441. This evidence implied that Professor Zannad considered that the trial had yielded correct results in relation to both endpoints. A positive result for the use of ularitide, on the population tested, for the endpoints tested, could not be expected to have been seen, and that, in Professor Zannad’s view, was mainly because of the nature of the product. Furthermore, Professor Zannad’s view was that the results in relation to ularitide were consistent with other negative studies ‘in this space’. He clearly considered that the TRUE-AHF study, taken with those other studies, had led to the consensus view amongst cardiologists moving away from the idea that ularitide and similar drugs would be effective in the treatment of ADHF.
442. These points lead me to conclude that it was not the fact of the enrolment of ineligible patients which led to the study being negative (in the sense of showing an effect size considerably less than that which would have been regarded as outweighing the adverse effects of the drug and as meriting regulatory approval and having a p-value which did not meet the pre-specified level). Instead, I find on the evidence adduced that the study was negative in that sense because that accurately reflected the effect of the drug on the population involved, for Endpoint 1, as well as for Endpoint 2.
Legal principles applicable to claims for wasted costs as damages
443. There was little dispute between the parties as to the law relating to claims for wasted costs.
444. The fundamental principle is that stated by Parke B in Robinson v Harman (1848) 1 Exch 850 at 855:
‘The rule of the common law is, that where a party sustains a loss by reason of a breach of contract, he is, so far as money can do it, to be placed in the same situation, with respect to damages, as if the contract had been performed.’
445. One way in which the claimant’s interest in the contract’s being performed is protected is by an award of damages in the amount of expenditure which the claimant has wasted as a result of the defendant’s breach of contract: see Anglia Television v Reed [1972] 1 QB 60; CCC Films Ltd v Impact Quadrant Films Ltd [1985] 1 QB 16; Commonwealth of Australia v Amann Aviation Pty Ltd (1991) 174 CLR 64 (HC of Australia).
446. In Anglia v Reed it was said that this claim was for expenditure ‘which has been thrown away, that is, wasted, by reason of the breach…’. In Galtrade v BP Oil [2021] EWHC 1796 (Comm), at [117] Adrian Beltrami QC, sitting as a High Court Judge, said:
‘It is important also to understand that the mere incurrence of expenditure does not in and of itself give rise to a loss sounding in damages. Under the so-called reliance measure, the critical further component, which converts the expenditure into a potentially recoverable loss, is that this has been wasted or rendered futile by reason of the breach.’
447. The burden of proving that expenditure is wasted is on the claimant. This was common ground, correctly in my view. The ordinary onus is on the claimant to prove its loss, and this is not an area in which it is necessary to apply any presumption of loss or reversal of the burden of proof in order to ensure justice to the claimant.
448. A claim for wasted expenditure is not an exception to the fundamental principle stated in Robinson v Harman, but is a method of giving effect to it. This is made clear in Omak Maritime Ltd v Mamola Challenger Shipping Co (The ‘Mamola Challenger’) [2011] 1 Lloyd’s Rep 47. It follows that the court will not knowingly allow a claim for wasted expenditure to put the claimant into a better position than it would have been had the contract been performed: The ‘Mamola Challenger’; Yam Seng Pte Ltd v International Trade Corporation Ltd [2013] EWHC 111 (QB) at [186]. This was summarised by Adrian Beltrami QC in Galtrade at [125]:
‘[A claim for wasted expenditure] has validity only if and to the extent that the Court is satisfied that the claimant would indeed have generated the necessary revenue to recoup the expenses. The Court will not knowingly make an award of damages which puts the claimant in a better position than if the contract had been performed and so will not allow a claim for wasted expenditure if the result of the breach is that the claimant has thereby managed to extricate itself from a loss-making transaction.’
449. However, once the claimant has shown that expenditure has been wasted as a result of the defendant’s breach, then it benefits from a rebuttable presumption that it would have generated enough revenue to at least break even: The ‘Mamola Challenger’ at [47]; Yam Seng at [187]-[190]; The Royal Devon and Exeter NHS Foundation Trust v ATOS IT Services UK Ltd [2017] EWHC 2197 (TCC) at [68]; Galtrade at [126-127].
450. In some cases, the benefits which the claimant intended to gain under the contract may not be financial, but alternative gains such as use or enjoyment. In such cases, the position is as set out in The Royal Devon and Exeter NHS Foundation Trust at [68] per O’Farrell J:
‘To establish a bad bargain in such a case, the defendant would have to show that the value of the asset or other performance promised was less than the expenditure incurred by the claimant.’
The assessment of waste and the relevance of benefits received
451. One issue which was the subject of some debate was the relevance of benefits which have been received by the claimant under the contract. The correct approach is, in my judgment, that to the extent that a claimant has received a benefit, it cannot, pro tanto, show that its expenditure has been wasted. If, after taking the benefit into account, expenditure can be seen to have been wasted, then that wasted expenditure can be recovered, subject to the defendant’s showing that the total benefit that the innocent party would have received under the contract if it had been performed would have been worth less than the expenditure incurred. That is an approach consistent with the decision in Grange v Quinn [2013] EWCA Civ 24, see especially paragraphs 86-87 per Jackson LJ and 128-129 per Gloster J.
452. It was submitted by Cardiorentis that the obiter remarks of Christopher Nugee QC (as he then was), sitting as a High Court Judge in Khan v Malik [2011] EWHC 1319 (Ch) at [129]-[132] are to the effect that, in a case in which the defaulting party has provided some part of the contractual performance, there cannot be a claim for recovery of wasted expenditure as damages, and that this was wrong. If those paragraphs said that, I would agree that that was incorrect. A claim for wasted expenditure is not dependent on a showing of a total failure of basis. I agree with the statement in Chitty on Contracts para. 32-069, to that effect. I regard the remarks of the judge in Khan v Malik as explicable on the basis that he was rejecting the argument that the claimant could recover the entire amount paid, even though some benefit had been received, and the judge was not considering an argument to the effect that there could be recovery of a lesser amount which took into account the value of such performance as there had been.
453. In most of the authorities in this area, there is little consideration of what constitutes wasted expenditure, for in them there has been generally been total, or near total non-performance and expenditure was clearly thrown away. The present case is one where, though Cardiorentis received the results of the TRUE-AHF study, it contends that the costs of obtaining those results were wasted because of IQVIA’s breaches. As already discussed, that claim was not and could not be put on the basis that the expenditure was wasted simply because the results were negative, but was put on the basis that the quality of the study and its results were such as to mean that the expenditure was wasted. The question was therefore, what quality would the study have had to have for the expenditure on it not to be regarded as wasted?
454. Cardiorentis’s pleaded case was that expenditure had been wasted because the study was of little scientific or practical value, and had not yielded data which could be used reliably for the purpose of providing Phase III data for submission to regulatory authorities and for licensing. In argument Cardiorentis suggested a formulation of the test for whether expenditure was wasted as being whether the study yielded an answer which satisfied the scientific community. It further made it clear that a test which satisfied the scientific community would satisfy the relevant regulators, there being no practical difference between the two. While a formulation in terms of acceptance by the ‘scientific community’ might be thought to be imprecise, as that community may not be entirely homogeneous, the evidence in this case indicated that it is a meaningful concept.
Application of legal principles to the facts
455. Applying these legal principles to my findings of fact gives rise to few difficulties.
456. I have concluded that Cardiorentis has not established that any material number of eligibility (or other protocol) deviations were caused by IQVIA’s breach or breaches of contract. It has accordingly not been shown that it was due to breach or breaches by IQVIA that, if the number or nature of eligibility violations caused difficulties in the interpretation of the study, this was the fault of IQVIA.
457. In any event, the results of the TRUE-AHF study were reliably negative, in the way I have described above. Those results were, as I have concluded, an accurate reflection of the effect of the drug on the population tested. Furthermore, those results, taken together with other evidence, were such as to have satisfied the scientific community as to the effectiveness or otherwise of ularitide in relation to Endpoints 1 and 2, in a population such as the one tested. In those circumstances, the expenditure on the study cannot have been said to have been wasted.
458. Because I have found that the study was reliable in relation to Endpoint 1 as well as Endpoint 2, the question does not arise as to how account is to be taken of the value of the negative findings which the study produced in relation to Endpoint 2, and which Cardiorentis did not dispute were reliable. I accept, however, that had I found that the study was unreliable in relation to Endpoint 1 by reason of IQVIA’s breach, it would have been relevant to have asked, as part of assessing the extent to which expenditure had been wasted, what value was attributable to reliable results in relation to Endpoint 2. I also consider that, for these purposes, the cost of obtaining the results in relation to Endpoint 2 can be taken as evidence of that value.
459. Here, had the issue arisen, I would have concluded that the cost of obtaining the results in relation to Endpoint 2, evidencing the value obtained, were all the costs of the study between February 2014, when Endpoint 2 was added, until November 2015, when the DSMB decided that Endpoint 2 should be abandoned for futility. Cardiorentis could not have obtained the results in relation to Endpoint 2 by spending less than the money expended on the study as a whole. I do not consider that it is possible to say that the costs of the trial during that period are to be treated as only referable to Endpoint 2 to the extent that they were attributable to the (limited) additional number of patients required to investigate Endpoint 2. During that period, all the investigation was designed to obtain results on both endpoints. Cardiorentis undoubtedly wanted to have the results in relation to Endpoint 2. If positive the results on Endpoint 2 would have added very significantly to the value of the drug. In 2014 Cardiorentis estimated that the revenues which might be generated if ularitide had a positive effect in relation to cardiac mortality had an NPV of US$16.492 billion. This may be compared with an estimated NPV of US$3.001 billion if it had an effect only in reducing time and treatment in hospital by preventing worsening clinical status, and of negative US$0.05 billion if it had an effect only on symptom relief.
460. As set out above, I have held that it has not been shown that there was any wasted expenditure. Had I concluded that there was wasted expenditure, the further issue would have arisen as to whether IQVIA had shown, the burden being on it, that the bargain would still have been a bad one, even had there been proper contractual performance.
461. In this regard, as set out above, the cases recognise that there may be some situations in which the relevant contract is not made with a view to financial gains. I do not consider that this case is one of them. Cardiorentis’s reason for sponsoring the study was because of its belief and hope that it would produce positive results, which would then allow ularitide to be approved for use, permitting significant sales and an increase in the value of Cardiorentis’s licence. This is illustrated vividly by a Partnering Presentation prepared by Cardiorentis and which was used for meetings with a number of pharmaceutical companies including Bristol-Myers Squibb in August 2014.
462. The question of whether the contract has non-financial gains in view is not to be answered by looking at it in isolation. The contracts in Anglia TV v Reed, CCC Films and other cases would not have gained a profit directly, but rather were facilitative, in that the contracting party acquired a chattel or right intended to be used to generate profits. That was the case here: it was intended that the results of the trial would allow approval and marketing of the drug.
463. The question, accordingly, is whether IQVIA could show that, if there had been proper performance of the contract with Cardiorentis, Cardiorentis’s gross profit from exploitation of its rights to develop ularitide would have at least allowed it to break even, after allowing for the costs of the Trial. The question arises in circumstances where I have found that, in any event, no causative breach has been made out. Nevertheless, I have considered whether, on any plausible view of the facts and the outcome of the study, assuming proper performance by IQVIA, Cardiorentis would have made a gross profit which covered the cost of the study. I consider that it is clear it would not have done so. This is because the results in relation to Endpoint 2 were negative; and because the results in relation to Endpoint 1 were so far from showing an effect size of the magnitude which the FDA had pre-agreed was necessary for the drug to be approved, that it seems clear that it would not have been. Thus, even if it could be said that there were some failures on the part of IQVIA, and they had some effect in making the results of the study less favourable to the drug than they would otherwise have been, they cannot realistically have made sufficient difference to obscure an effect size of the magnitude which had been discussed with the FDA and on which the study size in the Protocol was based. That this is so is supported not just by the results of the study itself, but by comparison with the ASCEND and RELAX-2 studies, and how the scientific community and pharmaceutical companies have reacted to the TRUE-AHF study and other studies in the area.
The Claim for an Injunction
464. Cardiorentis claims that it is entitled to an injunction that IQVIA should provide it with access to the data and information generated as a result of the TRUE-AHF trial, including the TMF database, and delivery up of all trial data, information and documents. The issue arises because in about May 2017 IQVIA terminated Cardiorentis’s access to the eTMF, and this has not been reinstated, and there has been no delivery up of the study materials or data.
465. In relation to delivery up of the study information, IQVIA’s case that that is subject, under clause 8, to payment under the contract, is not disputed by Cardiorentis. Accordingly, if Cardiorentis has not paid in full, it is not entitled to delivery up pursuant to clause 8.
466. Cardiorentis contends, however, that the position is different in relation to data access. It relies on the legislative framework against which the GSA was entered into, as relevant to its construction. That framework included that there should be a master file for study data, to which the Sponsor should have access. In England, for example, a trial Sponsor must, under reg 31A(2) Medicines for Human Use (Clinical Trials) Regulations 2004 (SI 2004/1031), keep a trial master file and must ensure that it is readily available at all reasonable times for inspection by the licensing authority or by any person appointed by the Sponsor to audit the arrangements for the trial. Clause 7 of the GSA enshrines Cardiorentis’s ownership of trial data. Cardiorentis contends that, consistently with these matters, it did have access to the eTMF during the trial, and that nothing in clause 8 allowed IQVIA to deny Cardiorentis access to its data pending satisfaction of payment conditions elsewhere in the contract. The consequence of Cardiorentis not having access to the eTMF is that it can impede or hamper Cardiorentis’s position with regulators. As Cardiorentis contends, it is clear that clause 8 is not intending to interfere with its access to the master file, not least because the last sentence of clause 8 confirmed that nothing in the GSA transfers from Cardiorentis to Quintiles any FDA or regulatory record-keeping requirements.
467. I have concluded that Cardiorentis is correct in relation to this. I consider that it is implicit in the GSA, taking into account the regulatory framework against which it was made, and which both parties knew of, that Cardiorentis should have access to the eTMF, containing what, by clause 7 of the GSA, was its data and information. Clause 8 does not, in my view, seek to regulate such access, but is concerned with a process of delivery up of other materials, information and data, which IQVIA retains at the conclusion of the services.
The Counterclaim
468. IQVIA counterclaims for amounts due under invoices issued in respect of work on the study, which it contends are unpaid. These were claimed in its POC in Action CL-2018-000841 in the sum of Eur 9,501,587.25, and interest. Cardiorentis pleaded a defence of set off of the damages it claimed on its own claim, and otherwise put IQVIA to proof of IQVIA’s entitlement to recover the sums claimed.
469. The principal evidence relied upon by IQVIA in support of its counterclaim was that of Mr Pauljit Hira. Mr Hira had produced a schedule of invoices which were considered outstanding, and which was appended to his witness statement. That schedule omitted four of the invoices which had been included in the Schedule to the POC in Action CL-2018-000841. Mr Hira confirmed in his oral evidence that his schedule was intended to be comprehensive, and he was cross-examined on that basis. No further evidence was called relevant to the recoverability of these invoices. In those circumstances I consider, as submitted by Cardiorentis, that IQVIA’s claim must be taken to be for an amount calculated omitting those four invoices, which totalled Eur 181,319.10.
470. In addition to this, during the trial, IQVIA accepted that the amount for which it could claim should be reduced by an amount of Eur 1,078,896, on the basis that some sums previously billed for as pass-through expenses fell within the scope of IQVIA’s budgeted fees.
471. There were specific issues in relation to two invoices, each of which related to a performance-related ‘milestone’ as set out in Change Order 7.
472. The first of those invoices was THV-158125, in an amount of Eur 2,332,579.80. This was based on IQVIA’s alleged entitlement to an amount of 5% of its undiscounted Traditional Services fee in relation to the milestone of ‘90% data clean’ by 29 February 2016. Cardiorentis put IQVIA to proof that 90% data clean had been achieved by that date. In answer to that IQVIA relied on three documents: first, a Weekly Progress Report circulated to Cardiorentis on 29 February 2016 which stated that by 26 February 2016, 99.95% of forms had been SDVed, 94% of patients had had all pages SDVed, and 95% of patients were clean up to day 30; secondly an email from Dr Spaeder to Dr Holzmeister on 30 March 2016 recording a conversation in which Dr Holzmeister said that he planned to provide an email confirming that IQVIA had achieved the data cleaning milestone; and third an email from Mr Wittwer to IQVIA dated 31 March 2016 confirming that the 90% data cleaning milestone had been met.
473. IQVIA did not plead that the communications from Dr Holzmeister on 30 March 2016 or Mr Wittwer on 31 March 2016 had a contractual or estopping effect. The question therefore remains whether, on the evidence available it is shown that IQVIA had met the ‘90% data clean’ milestone. It points to the 26 February 2016 Weekly Progress Report, but that is not specifically directed to whether the milestone has been met. For example, while the proportion of patients said to be ‘clean’ is 95%, that is up to Day 30, and there is no indication as to what quantity of data might relate to days after Day 30 which is not clean; nor is there an indication of the amount of ‘uncleaned’ data which was involved in the 5% of patients who were not ‘clean’. That Weekly Progress Report has to be considered together with: (1) an email form Ms Queiroz to CRAs on 22 February 2016, which had indicated that there was still a lot to do by 1 March 2016 ‘in terms of monitoring and data cleaning’, and had indicated that in the case of 72 patients there were open queries, 33 patients were ‘To SDV’, and 233 ‘To SDV/To Freeze’; and (2) an email from Ms Ron of 23 February 2016 which had said, commenting on Ms Queiroz’s email, that ‘the data cleaning status is not good’.
474. The material which IQVIA adduced in relation to this issue was markedly less extensive and detailed than I would have expected given the depth and thoroughness of the investigation of the facts relating to the trial which has occurred in this action. The material which has been produced leaves me uncertain as to whether ‘90% data clean’ was achieved by 29 February 2016, and I find that IQVIA has not proved its case on this point on the balance of probabilities.
475. The other specific invoice about which there was debate was invoice THV-159368, relating to the milestone for ‘Statistical Analysis - Top Line Result’ delivery, in the amount of Eur 1,399,547.88. The issue here is whether IQVIA met the milestone by providing ‘statistical analysis - top line result’ within 13 days after Database Lock. Cardiorentis contended that it did not; IQVIA said that it did.
476. A series of TLFs, including Table 14.2.1.1, was provided by IQVIA to Cardiorentis on 24 March 2016, within 13 business days of Database Lock. On 19 April 2016, outside the 13 day period, IQVIA sent an updated version of these TLFs, which corrected various figures. The amendments included some changes to Table 14.2.1.1. Thus, the number of patients on ularitide said to have ‘worsened’ was changed from 72.3 (6.6%) to 70.7 (6.5%), and on placebo from 88.7 (8.3%) to 87.6 (8.2%); while the p-value was changed from 0.830 to 0.824.
477. IQVIA contends that whether it was entitled to the milestone payment had to be capable of being known when the results were submitted, and that what had occurred on 24 March 2016 constituted delivery of the top line result. Alternatively, IQVIA submitted that none of the changes changed the result, and led to a de minimis change to the p-value. Cardiorentis contended that any correction to the numbers in the main results table outside the 13 day period meant that what had been provided within that period was not the delivery of the ‘top line result’. Alternatively it submitted that if the changes were material, then what had been submitted was not the ‘top line result’, and here the changes were material, in that they affected the overall results of the study.
478. The question is one of construction, bearing in mind the background to and purpose of the provision. That purpose was to provide an incentive payment (by way of ‘earn back’ of a discount) if IQVIA completed an aspect of its performance within a certain period. In my judgment, it was an integral part of that purpose that the result provided should accurately reflect the results of the trial. The parties were clearly not agreeing that the incentive payment should be made if something which could be called a topline result was submitted within 13 days, however inaccurate a reflection of the results of the study.
479. The question is whether any deviation from an accurate statement of the results of the study means that the result provided did not constitute ‘the top line result’. While there is a superficial attraction in saying that the departure must be material for the results provided not to constitute the top line result, it is difficult to say precisely what that might mean. The context is of biostatistics in relation to a clinical study: accuracy is clearly of great importance. While the change in the p-value here was only 0.006, it is not difficult to think of situations where that difference might have been of potential importance: for example if the results had shown a p-value of 0.013 as against 0.007, or, in the context of the most usual identifier of statistical significance, 0.053 as opposed to 0.047. In the present case, of course, given the results, the difference between 0.830 and 0.824 was not of particular importance, but I do not consider that the question as to what degree of divergence would mean that there was not a ‘top line result’ for the purposes of Change Order 7 is answered by reference to questions of whether the overall results of the trial would be different from the perspective of whether the drug would obtain regulatory approval, or be or a commercial success, or similar considerations.
480. I conclude that the right construction of the Change Order 7 is that IQVIA had to present accurate figures within the 13 days to gain the performance bonus. If it had to amend the figures subsequently, that would reflect that it was not in fact ready to provide such figures within the 13 days, and thus not in a position to earn the performance bonus. Accordingly I consider that Cardiorentis is correct in relation to this issue as well.
481. I considered that IQVIA had established on a balance of probabilities, and in the absence of specific issues having been raised in relation to the work / expenses covered by them, that it is entitled to recover in respect of the invoices in Mr Hira’s Schedule other than those I have dealt with above.
482. In the circumstances, IQVIA is entitled to succeed in a principal amount of Eur 4,509,244.47. I will hear further submissions as to what if any interest should be paid in relation to that amount.
Overall Conclusion
483. For the reasons given above, I find:
(1) Cardiorentis’s claim for damages fails;
(2) Cardiorentis’s claim that it is entitled to an injunction that it should have access to data relating to the TRUE-AHF study succeeds;
(3) IQVIA is entitled to succeed in relation to its counterclaim in an amount of Eur 4,509,244.47.