MR JUSTICE ARNOLD :
Contents
| Topic |
Paragraphs |
| |
|
| Introduction |
1-7 |
| Technical background |
8-53 |
| MS |
9-22 |
| MRI |
23-26 |
| Use of MRI in MS |
27-29 |
| Use of disability scores |
30 |
| Causes of MS |
31-34 |
| MS therapies |
35 |
| Disease-modifying therapies |
36-39 |
| Self-injection |
40 |
| Injection-related side effects |
41 |
| Clinical trials of GA |
44 |
| Bornstein 1987 |
45 |
| Johnson 1995 |
46 |
| Comi 2001 |
47 |
| Filippi 2006 (CORAL) |
49 |
| Cohen 2007 (FORTE Phase II) |
50 |
| COMI 2008 (FORTE Phase III) |
51 |
| Mode of action of GA |
52 |
| Pharmacokinetics of GA |
53 |
| The Patent |
54-71 |
| Background to the invention |
55-56 |
| Summary of the invention |
57-58 |
| Detailed description of the invention |
59 |
| Definitions |
60-61 |
| Experimental details |
62-65 |
| Results |
66-68 |
| Discussion |
69-71 |
| The claims |
72-74 |
| The skilled person |
75-78 |
| The expert witnesses |
79-93 |
| Common general knowledge |
94-133 |
| US versus UK Knowledge |
98-99 |
| Side effects of the DMTs |
100-101 |
| Adherence and convenience |
102-111 |
| The 20 mg QD regimen for GA administration |
112 |
| The FORTE trials |
113-127 |
| Work on lower frequency administration of GA |
128-132 |
| New treatments on the horizon |
133 |
| Claim interpretation |
134-144 |
| The law |
134-139 |
| Claim 1 |
140-143 |
| Claim 3 |
144 |
| The prior art |
145-156 |
| Pinchasi |
146-151 |
| Caon |
152-153 |
| Flechter |
154-156 |
| Novelty |
157-173 |
| The law |
157-167 |
| Novelty over Pinchasi |
168-173 |
| Claim 1 |
168-172 |
| Claim 3 |
173 |
| Obviousness over the prior art |
174-190 |
| The law |
174 |
| Obviousness over Pinchasi |
175 |
| Claim 1 |
175-184 |
| Claim 3 |
185 |
| Secondary evidence |
186-188 |
| Caon and Flechter |
190 |
| Lack of inventive step for want of technical contribution and insufficiency |
191-200 |
| Arrow declaration |
200-212 |
| The law |
201-203 |
| Assessment |
204-212 |
| Summary of conclusions |
213 |
Introduction
- The Claimants ("Mylan" and "Synthon") seek revocation of European Patent (UK) No. 2 949 335 entitled "Low frequency glatiramer acetate therapy" ("the Patent") of which the Defendant ("Yeda") is the registered proprietor and the Third Party ("Teva") is the exclusive licensee. In addition, the Claimants seek an Arrow declaration. I will refer to the Defendant and the Third Party collectively as "the Defendants". The Defendants counterclaim for threatened infringement of the Patent.
- This is the third round of proceedings in this Court between the parties, or some of them, concerning glatiramer acetate ("GA"). The first concerned European Patent (UK) No. 0 762 888 ("888"), the basic patent protecting low molecular weight GA. Mylan applied to revoke 888 and sought a declaration of non-infringement. I rejected the attack on the validity of 888 and refused a declaration of non-infringement ([2012] EWHC 1848 (Pat)) and the Court of Appeal dismissed Mylan's appeal ([2013] EWCA Civ 925, [2014] RPC 4). 888 expired on 23 May 2015.
- The second round concerned European Patents (UK) Nos. 2 177 528 and 2 361 924 ("924"), which concerned improved processes for the preparation of GA. Synthon applied to revoke both patents. Birss J rejected the attacks on the validity of both patents with the exception of claims 20, 27 and 28 of 924. The Court of Appeal dismissed both Synthon's and Teva's appeals with respect to 924 ([2017] EWCA Civ 148) and the Supreme Court refused Synthon permission to appeal. On 12 September 2017, however, a Technical Board of Appeal of the European Patent Office revoked 924 in its entirety.
- The Patent is directed to a dosage regimen for the administration of GA for the treatment of relapsing forms of multiple sclerosis ("MS") consisting of three subcutaneous injections of 40 mg GA every seven days with at least one day between each injection ("40 mg TIW"). Previously, GA was approved for administration in a regimen consisting of a daily subcutaneous injection of 20 mg ("20 mg QD"). The Claimants contend that the Patent is invalid on the grounds of lack of novelty, lack of inventive step and insufficiency. There is no challenge to the earliest claimed priority date of 20 August 2009.
- A fourth round of proceedings is scheduled for trial in April 2018. That concerns European Patent (UK) No. 3,050,556, which is another patent for an improved process for the preparation of GA.
- The reason for the current litigation is not hard to find. Teva markets GA under the trade mark Copaxone. Worldwide sales of Copaxone in the year ending 31 December 2016 were about $4.2 billion, representing nearly a fifth of Teva's worldwide sales and a significantly higher percentage of its profits. Since GA has been authorised for administration in accordance with the 40 mg TIW regimen, a large proportion of prescriptions of GA has been written for that regimen. The Claimants have previously introduced a 20 mg GA generic product. Now they wish to clear the way for the launch of a 40 mg GA generic product for which they obtained a marketing authorisation on 5 October 2017 ("the Claimants' Product"). There is no dispute that, if the Patent is valid, the Claimants' intended acts in relation to the Claimants' Product would infringe it.
- One measure of the importance of the case to the parties is provided by the fact that the Claimants' closing written submissions extend to 191 paragraphs (plus three annexes) while the Defendants' closing written submissions extend to 306 paragraphs (plus one annexe) even though the issues are, with one exception, fairly straightforward. I have taken all these submissions into account, but it is not necessary or appropriate for me to address every single one in this judgment.
Technical background
- Most of the following account of the technical background is based on the primer which was sensibly agreed between the parties. Like the primer, it is expressed in the present tense for convenience, but records the position as at August 2009. I have added sections on clinical trials, injection-related side effects and the pharmacokinetics of GA based on the expert evidence which could usefully have been included in the primer.
MS
- MS is a chronic, disabling, neurodegenerative autoimmune disease of the central nervous system ("CNS"). Because diagnosis often coincides with individuals beginning or building a family, as well as with the time of life that may include critical career decisions, education or training, MS can be particularly devastating to family, social and professional relationships as well as adversely affecting a patient's ability to work.
- MS is an autoimmune inflammatory condition in which the body's immune system attacks elements of the CNS, namely the insulating myelin sheath in the brain, optic nerve and spinal cord. Immune cells from the periphery enter the CNS and those cells (including lymphocytes) of the immune system mount an immune response against elements of myelin. Myelin is an insulating substance constituted by the cell membrane of a specialised cell known as the oligodendrocyte that surrounds nerve fibres. The myelin sheath is essential for the efficient conduction of nerve signals and provides trophic support to maintain the survival of nerve fibres. In patients affected by MS, the immune-mediated breakdown of myelin ("demyelination") interferes with the conduction of nerve signals and leads to abnormalities including, but not limited to, motor, sensory, cognitive, visual and sphincter-based neurological dysfunction.
- Although full details of the cause or causes of MS remain unknown, a tremendous amount about the disease and how the immune system behaves in the CNS has been learned and is now known. There is a significant genetic contribution, and environmental factors also appear to play a role (people living in cold temperate climates are at increased risk which may be due to low vitamin D levels). The timing of certain viral infections may play a role in triggering the onset of MS in susceptible individuals.
- Demyelination may occur at various sites within the CNS, affecting movement and sensory functions, as well as in the optic nerves, affecting vision. The symptoms experienced by a patient depend upon the site or sites within the CNS that are affected by demyelination and by the size of the lesions.
- Characteristic clinical manifestations of MS include: motor weakness; partial paralysis of the limbs; prickling or tingling sensations on the skin; loss of awareness of the body's position in space; blurred vision or loss of sight; loss of muscle coordination; walking difficulties; cognitive problems and bladder dysfunction.
- MS is a chronic condition of varying severity. Some MS patients are minimally affected by the disease, while other affected patients progress relatively swiftly to significant levels of disability. Although every individual will experience a different combination of MS symptoms, a number of distinct patterns relating to the course of the disease are recognised.
- The most common form of MS is relapsing-remitting MS ("RRMS"). More than 80% of people diagnosed with MS have RRMS at the time of diagnosis. Patients with RRMS experience intermittent, unpredictable relapses, characterised by the appearance of new symptoms and/or increased severity of existing symptoms typically worsening over a period of hours to days. Relapses also last for varying periods (days, weeks or in some cases months) and are followed by partial or total remission. The time between relapses also varies. For some patients with RRMS, their disease may be clinically inactive for months or even years at a time with no evident relapses or apparent worsening of their disease.
- After a variable period of time, many patients with RRMS progress to a relentlessly debilitating form of the disease, known as secondary progressive MS ("SPMS"). Patients with secondary progressive MS demonstrate insidious or aggressive neurological deterioration with functional impairment becoming more severe over time. There is no "bright line" for the transition between the relapsing-remitting and secondary progressive forms of MS. Some patients with SPMS experience occasional superimposed relapses, with intervening partial recovery of function, but this is the exception rather than the rule.
- A number of less common variations in the clinical presentation of MS have also been described. These include primary progressive MS ("PPMS"), characterised by a steady progression of clinical symptoms and increasing disability without distinct relapses or remissions from onset, and relapsing progressive MS ("RPMS"), characterised by a gradual progression from the onset of the disease accompanied by occasional episodes in which the severity of symptoms increases. A diagram comparing the different forms of MS showing increase in disability against time is set out below.
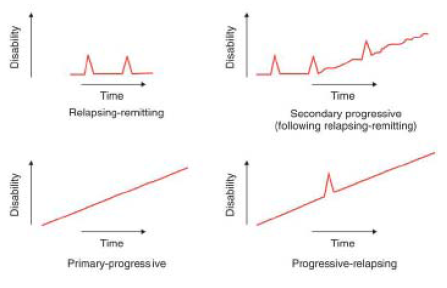
- In addition, if patients are identified at the time of their first clinical attack of disease and thus do not (yet) have evidence of disease recurrence to support a diagnosis of MS, they may be classified as having clinically isolated syndrome ("CIS"). This is characterised by a single episode of clinical symptoms. Most of these patients go on to develop RRMS.
Diagnosis of MS
- There are established criteria used to diagnose MS. These include the older Poser criteria and the more recent McDonald criteria.
- The Poser criteria initially established that, to make a diagnosis of "clinically definite MS", it was necessary to demonstrate that the disease involved more than one pathway (dissemination in space) and was not monophasic (dissemination in time). This method relied on clinical evaluation, and typically required that a relapsing patient have two identified attacks (i.e. relapses) of more than 24 hours duration separated by an interval of more than one month together with clinical evidence of lesions in at least two different places within the CNS. The Poser criteria were dependent upon neurological examination.
- Under the McDonald criteria, which were first introduced in 2001 and then revised in 2005, in addition to characteristic symptoms and signs on neurological examination, patients can be diagnosed based on characteristic findings on magnetic resonance imaging ("MRI") of the brain and spinal cord. Where a patient presents with a history of two neurological episodes characteristic of MS, but, upon examination, only one lesion is confirmed by objective clinical evidence, the McDonald criteria (as revised in 2005) permit MRI findings to be relied upon to establish that the disease has affected more than one area of the CNS and to support a diagnosis of MS. Similarly, where neurological examination reveals objective clinical evidence of two or more lesions in the CNS, but the patient history suggests only one attack, the McDonald criteria (as revised in 2005) permit MRI findings to be relied upon to establish that the disease process was disseminated in time to make a diagnosis of MS.
- Patients presenting with CIS typically have experienced a single episode of symptoms consistent with MS along with a finding of a single neurological lesion upon examination. In addition, MRI scans of such patients, whilst consistent with MS, do not fulfil the McDonald criteria to enable a firm diagnosis of MS.
MRI
- MRI is used in MS in (i) the diagnosis (using the McDonald criteria), (ii) tracking the progress of the disease and (iii) measuring the response of patients to treatment. MRI is used extensively in neurology and neurosurgery because it provides exquisite structural evaluation of brain, spinal cord and vascular anatomy in three dimensions typically divided into different planes of cross section (axial, coronal and sagittal).
- MRI is based on the magnetisation properties of atomic nuclei, in particular protons, which are present in the water of living tissue. A proton has an electro-magnetic field around it, and precesses ("spins") around its own axis. Normally, the electro-magnetic fields of protons within tissue are randomly oriented. In MRI, a powerful, external magnetic field is applied to align the nuclear spins of the protons within the particular slice of tissue being examined. Once the nuclear spins of the protons have been aligned, a pulse of radio-frequency energy is introduced which disrupts the alignment of the nuclear spins of the protons as the protons absorb the energy of the radio-frequency pulse. When the pulse is turned off, the nuclei return to their resting alignment through various relaxation processes and the radio-frequency energy absorbed during the pulse is re-emitted.
- The amount of radio-frequency energy that is absorbed and subsequently emitted on relaxation, as well as the time taken for relaxation to occur, depends upon the environment and chemical nature of the proton. For example, a proton located within the cerebrospinal fluid ("CSF") will behave differently to one located within the white matter. By measuring the radio-frequency energy emitted and the time taken for relaxation, it is possible to produce a picture of the imaged slice of the brain, in which different tissues (e.g. grey and white matter or injured tissue) are differentiated. Low molecular weight gadolinium ("Gd") complexes are paramagnetic contrast media that are sometimes administered to patients prior to MRI to identify areas of disruption of the blood-brain barrier or highly vascularised lesions.
- Tissues can be characterised by two different relaxation times: T1 and T2. These relate to the two different relaxation behaviours that occur when the radio-frequency pulse comes to an end: T1 is the longitudinal relaxation time and T2 is the transverse relaxation time. Liquids, such as CSF, have longer T1 and shorter T2s, whereas fats, such as white matter, have shorter T1 and longer T2s. Hence it is possible to create a differentiated image of the brain utilising the measurements of T1 and T2.
Use of MRI in MS
- There are a number of different types of MRI scans that are useful in the diagnosis, monitoring and tracking of MS, in particular T1- and T2- weighted images:
i) new, active sites of blood-brain barrier breakdown, and thus inflammatory demyelination within the CNS, may be visualised as Gd-enhancing areas on T1-weighted images;
ii) new MS lesions may also be visualised as hyperintense areas (i.e. bright spots) on T2-weighted images; and
iii) old lesions may appear as hypointense areas (i.e. black holes) on T1-weighted images.
- Analysis of MRI scans include monitoring the number, location and volume of:
i) Gd-enhancing lesions on T1-weighted images;
ii) hyperintense lesions on T2-weighted images (new or enlarging); and
iii) hypointense lesions on T1-weighted images.
- Brain atrophy is a reduction in brain volume over time and is a marker of neurodegeneration and progression of disability. Changes in brain volume can also be assessed by MRI.
Use of disability scores
- The progress of MS in a particular patient is also monitored through the use of formal measures of disability. A common score is the Expanded Disability Status Scale ("EDSS"), also known as the Kurtzke Disability Status Score. Other commonly used scores are the Ambulation Index ("AI"), the Multiple Sclerosis Functional Composite ("MSFC") and the EuroQoL (or "EQ-5D") questionnaire.
Causes of MS
- While the precise origins of MS are not fully understood, researchers generally agree that inappropriate immune activation targeting the CNS followed by a breach in the integrity of the blood-brain barrier in susceptible individuals contributes to a cascade of events resulting in demyelination of CNS nerve fibres (axons) and the formation of lesions in the brain. Without the myelin sheath surrounding the axons, the ability to facilitate transmission of nerve impulses in the CNS is impaired.
- The autoimmune character of MS is supported by the presence of activated T-cells within MS lesions, the overwhelming contribution of genes involved in immune system function in genetic susceptibility to the disease and the therapeutic efficacy of immunomodulatory drugs (see below), in particular those that block the migration or infiltration of reactive T-cells into the CNS.
- There are studies indicating that certain genes, in particular HLA-DR1501, belonging to the Major Histocompatibility Complex ("MHC") class II category are strongly associated with genetic susceptibility to MS. MHC II refers to cell surface proteins found on antigen-presenting cells which play a role in binding peptides to facilitate their display to T-cells. Variation in MHC II can therefore have an impact on the immune response initiated by T-cells. Almost all of the genes identified to date are involved in immune system regulation or function.
- The development of autoreactive lymphocytes (particularly T-cells) with specificity for myelin antigens is therefore thought to play a central role in the initiation and continued disease activity of MS in patients.
MS therapies
- Although relapsing forms of MS are incurable, they are treatable in some patients. There are three general approaches to the attempted treatment of MS patients:
i) treatment with corticosteroids to accelerate recovery from acute relapses;
ii) treatment with disease-modifying agents, i.e. prophylactic therapy which reduces the number of relapses experienced by patients over a period of time and slows down, or hopefully prevents, long-term progression of the disease; and
iii) treatment of existing symptoms caused by prior injury, including fatigue, pain, muscle spasticity and visual deficits in order to relieve a variety of symptoms in the patient.
Disease-modifying therapies
- Disease-modifying therapies ("DMTs") for MS are therapeutic agents that work to suppress or modify the underlying immune dysfunction in patients. The goal of DMTs is to act as prophylactics to reduce or decrease the incidence of relapsing events, reduce inflammatory injury to the brain and thereby slow the progression of the disease.
- DMTs can reduce the frequency of relapses experienced by patients suffering from RRMS. In addition, such therapies are likely to be effective for slowing down progression of CIS to RRMS or RRMS to SPMS. All of the DMTs that are effective in changing the clinical course of MS are thought to act on the immune system.
- The following DMTs have been approved for use in the UK and are available for treating patients:
i) Interferon beta-1a (Avonex). Interferon beta-1a was first approved in the EU in March 1997 and has been available for use on the National Health Service ("NHS") since 2002 under the Department of Health's Risk-sharing Scheme (a scheme established by the Department to evaluate the long-term cost-effectiveness of DMTs after the National Institute for Clinical Excellence had concluded that it was not possible to evaluate their cost-effectiveness from the clinical trial data which was then available; the scheme enabled the price paid by the NHS to be adjusted at two year intervals to ensure cost-effectiveness). Interferon beta-1a is indicated for treatment of patients with relapsing forms of MS. Interferon beta-1a is administered by intramuscular injection once per week.
ii) Interferon beta-1a (Rebif). This form of interferon beta-1a was first approved in the EU in May 1998 and has been available for use on the NHS since 2002 under the Risk-sharing Scheme. This form of interferon beta-1a is indicated for treatment of patients with relapsing forms of MS. This form of interferon beta-1a is administered via subcutaneous injection three times per week as either a 22 µg or 44 µg dose, and the doses are administered, if possible, on the same three days each week (at least 48 hours apart).
iii) Interferon beta-1b (Betaferon). Interferon beta-1b was first approved in the EU in November 1996 and has been available for use on the NHS since 2002 under the Risk-sharing Scheme. Interferon beta-1b is indicated for treatment of patients with RRMS. Interferon beta-1b is administered via subcutaneous injection every other day ("QOD").
iv) Interferon beta-1b (Extavia). This form of interferon beta-1b was first approved in the EU in May 2008. It is indicated for treatment of patients with relapsing forms of MS and is identical to Betaferon. It is administered by subcutaneous injection QOD.
v) Glatiramer acetate (Copaxone). GA was first approved in the UK in August 2000 and has been available for use on the NHS since 2002 under the Risk-sharing Scheme. GA consists of the acetate salts of synthetic polypeptides containing four naturally-occurring amino acids: L-glutamic acid, L-alanine, L-tyrosine and L-Lysine with an average molar ratio of 0.141, 0.427, 0.095 and 0.338 respectively. The average molecular weight of GA is between 5,000 and 9,000 daltons. GA is indicated for treatment of patients with relapsing forms of MS. The recommended dose of GA in 2009 was 20 mg QD administered by subcutaneous injection.
vi) Natalizumab (Tysabri). Natalizumab was first approved in the EU in June 2006 and was made available for use on the NHS since 2007. Natalizumab is a recombinant humanised IgG4 monoclonal antibody and contains human framework regions and the complementarity-determining regions of a murine antibody that binds to α-4 integrin. This blocks entry of lymphocytes into the central nervous system across the vascular endothelial surface. Natalizumab is indicated for treatment of patients with relapsing forms of MS and also for patients with Crohn's disease. The recommended dose for natalizumab is 300 mg administered intravenously every four weeks.
- Clinical studies have shown that DMTs directly affect several MRI markers, for example, the number and volume of Gd-positive lesions, the volume of T1-hypointense lesions, the number of new or enlarging T2 lesions and the volume of T1-hypointense lesions. Reduction in brain atrophy is known to be another indicator of treatment success with DMTs.
Self-injection
- The treatments listed in paragraphs 38(i) to (v) above are administered by self-injection. Patients are instructed in self-injection techniques in advance of starting treatment. Sites for subcutaneous self-injection include the abdomen, arms, hips and thighs.
Injection-related side effects
- Subcutaneously injected DMTs such as interferon beta and GA have well known side effects associated with the injections, namely injection site reactions ("ISRs") and immediate post-injection reactions ("IPIRs").
- ISRs are common in patients injecting DMTs subcutaneously. A high proportion of patients experience injection site discomfort, reddening or fibrosis, and in some cases lipoatrophy. Lipoatrophy, or the localised loss of fat tissue, is disfiguring and permanent and can have a significant psychological impact on patients. ISRs have long been recognised as being associated with the frequency of subcutaneous injections that a patient administers to a particular site of the body. Accordingly, patients are instructed to rotate around different injection sites.
- In addition, some patients treated with subcutaneous DMTs experience an IPIR. This reaction typically presents as a constellation of certain symptoms, such as flushing, vasodilation, chest pain, dyspnoea, palpitations or tachycardia, immediately following injection of the drug. The timing, frequency and incidence of such IPIRs is not predictable, although they are always associated with an injection.
Clinical trials of GA
- There have been a number of clinical trials of GA over the years, including the following ones. It should be noted that, whereas early trials of GA (then called copolymer-1 or cop 1), including Bornstein 1987, involved the use of a material with a molecular weight in the region of 14,000 - 23,000 Daltons, Johnson 1995 and the other later trials involved use of a material with a lower molecular weight.
- Bornstein 1987. This was a single-centre, double-blind, randomised, placebo-controlled trial involving 50 patients suffering from RRMS. It was reported in Bornstein et al, "A pilot trial of Cop 1 in exacerbating-remitting multiple sclerosis", N. Eng. J. Med., 1987, 317(7), 408-414. It provided evidence of efficacy and safety of 20 mg QD GA in the treatment of this form of the disease.
- Johnson 1995. This was a multi-centre, double-blind, randomised, placebo-controlled Phase III trial involving 251 patients diagnosed with RRMS. The results were reported in Johnson et al, "Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind, placebo-controlled trial", Neurology, 1995, 45(7), 1268-1276 ("Johnson 1995"). Treatment with 20 mg QD GA resulted in a significant reduction in relapse rate and a reduced chance of worsening disability.
- Comi 2001. This was a multi-centre, double-blind, randomised, placebo-controlled Phase III trial involving 239 patients with RRMS. The results were published in Comi et al, "European/Canadian multicenter, double-blind, randomized, placebo-controlled study of the effects on glatiramer acetate on magnetic resonance imaging-measured disease activity and burden in patients with relapsing-remitting multiple sclerosis", Ann. Neur., 2001, 149(3), 290-297. They demonstrated that 20 mg QD GA significantly reduced MRI-measured disease activity and burden.
- Bornstein 1987, Johnson 1995 and Comi 2001 are collectively referred to as the "pivotal" studies since they provided the basis for GA's approval as a treatment for MS (although the US Food and Drug Administration approved it in 1996 on the basis of Bornstein 1987 and Johnson 1995).
- Filippi 2006 (also known as CORAL). This was a multi-centre, double-blind, randomised Phase III trial of 5 mg and 50 mg GA QD administered orally in 1651 patients with RRMS. The results were reported in Filippi et al, "Effects of oral glatiramer acetate on clinical and MRI-monitored disease activity in patients with relapsing multiple sclerosis: a multicentre, double-blind, randomised, placebo-controlled study", Lancet Neurol., 5, 213-220 (2006). The trial was a failure, in that it found that neither 5 mg nor 50 mg GA QD administered orally reduced the relapse rate or other clinical and MRI parameters of disease activity and burden.
- Cohen 2007 (also known as FORTE (FORTy mg Efficacy) Phase II). This was a double-blind, randomised Phase II trial comparing a 40 mg QD dosage regimen of GA with the 20 mg QD dosage regimen in reducing MRI activity and clinical relapses in 90 patients with RRMS. The results were reported in Cohen et al, "Randomized, double-blind, dose-comparison study assessing safety and efficacy of 40mg vs 20mg of glatiramer acetate on MRI-measured disease activity in relapsing-remitting multiple sclerosis", Neurology, 68(12), 939-944 (2007). I shall consider them below.
- Comi 2008 (also known as FORTE Phase III). This was a multi-centre, double-blind, randomised Phase III trial comparing the 40 mg QD regimen of GA with the 20 mg QD regimen in reducing MRI activity and clinical relapses in 1155 patients with RRMS. Teva announced the headline results in a press release dated 7 July 2008 which stated that "[t]he 40mg dose did not demonstrate increased efficacy in reducing relapse rate; however, the higher dose maintained the favourable safety and tolerability profile of COPAXONE® 20mg". The results were presented by Professor Giancarlo Comi, a world leader in the treatment of MS, at the World Congress on Treatment and Research in Multiple Sclerosis ("WCTRIMS") in Montreal in September 2008, although the results were not fully reported until after the priority date of the Patent. The abstract for Prof Comi's presentation was published in a supplement to the journal Multiple Sclerosis (Comi et al, "Results from a phase III, on-year, randomized, double-bind, parallel group, dose-comparison study with glatiramer acetate in relapsing-remitting multiple sclerosis", Multiple Sclerosis, 14, S299 (2008), "Comi 2008A") and Prof Comi's slides were published (and remain available) on the WCTRIMS website ("Comi 2008B"). Again, I shall consider the results below.
Mode of action of GA
- Whilst its precise mechanism of action is unknown, there are a number of theories as to how GA works to alleviate the symptoms of MS and/or to slow down progression of the disease. These include:
i) GA administration includes a shift from Th1 to Th2 type immune response in both human and animal subjects;
ii) experiments in animal model known as experimental autoimmune encephalomyeltiies ("EAE") have shown that GA specific Th2 cells accumulate in the CNS and release anti-inflammatory cytokines in a process known as bystander suppression;
iii) non-neutralising anti-GA immunoglobulins have been suggested by some to have a therapeutic benefit;
iv) GA administration induces a broad antigen non-specific alteration of APC function and interferes with antigen presentation via MHC class II molecules;
v) GA administration results in an increase in T-regulatory cells (CD4+/CD25+ Tregs);
vi) GA administration induces GA specific CD8+ suppressor T-cells; and
vii) GA might exert a direct neuroprotective effect.
Pharmacokinetics of GA
- An unusual feature of GA is that it is rapidly degraded into smaller peptide fragments soon after subcutaneous injection. It is not possible to detect GA in the bloodstream after administration. As a result, unlike most small molecule therapies, it is not possible to measure the pharmacokinetics of GA in humans. For this reason, and because there are no animal models that can predict dosing for GA, the only way to determine what doses or dosing frequencies are effective, safe and tolerable is by a trial in humans.
The Patent
- I will summarise the disclosure of the Patent using the same headings and sub-headings as those appearing in the specification.
Background to the invention
- The specification begins at [0001]-[0008] with a description of MS and its various forms, including RRMS and PRMS. At [0009]-[0010] it describes GA. At [0011] it records that Copaxone 20 mg daily injection is approved for the treatment of RRMS including CIS. At [0013] it refers to the Comi 2001 clinical trial, and at [0014] it states that data accumulated in clinical trials show that GA is "safe and well tolerated".
- At [0015] the specification states:
"Disclosed is an effective low frequency dosage regimen of GA administration to patients suffering from a relapsing form of multiple sclerosis, including patients who have experienced a first clinical episode and have MRI features consistent with multiple sclerosis"
Summary of the invention
- The specification then states:
"[0016] The invention is as defined in the appended claims. Described herein are methods of alleviating a symptom of relapsing-remitting multiple sclerosis (treating relapsing multiple sclerosis) in a human patient suffering from relapsing-remitting multiple sclerosis or a patient who has experienced a first clinical episode and is determined to be at high risk of developing clinically definite multiple sclerosis comprising administering to the human patient three subcutaneous injections of a therapeutically effective dose of glatiramer acetate over a period of seven days with at least one day between every subcutaneous injection so as to thereby alleviate the symptom of the patient.
[0017] Also described are methods of increasing the tolerability of GA treatment in a human patient suffering from relapsing-remitting multiple sclerosis or a patient who has experienced a first clinical episode and is determined to be at high risk of developing clinically definite multiple sclerosis which comprises reducing the frequency of subcutaneous injections of a pharmaceutical composition comprising a therapeutically effective dose of glatiramer acetate to three times over a period of seven days with at least one day between every injection"
- It may be noted that there is no reference in these paragraphs, or in [0018], to a 40 mg dose. Only at [0019]-[0022] does one find consistory paragraphs corresponding to the claims.
Detailed description of the invention
- At [0023]-[0063] the specification describes a considerable number of embodiments of the invention. The following passages are of note:
"[0024] In accordance with the invention possible injection schedules include Day 1, Day 3, Day 5; Day 1, Day 3, Day 6; Day 1, Day 4, Day 6; Day 2, Day 4, Day 6; Day 2, Day 4, Day 7; Day 2, Day 5, Day 7; or Day 3, Day 5, Day 7.
[0042] In yet another embodiment, the therapeutically effective dose of glatiramer acetate is 40mg/ml. In a further embodiment, the therapeutically effective dose of glatiramer acetate is 40mg/0.75ml.
[0049] In another embodiment, increasing the tolerability of GA treatment in the human patient suffering from a relapsing form of multiple sclerosis comprises reducing the frequency of an immediate post injection reaction.
[0051] In an additional embodiment, increasing the tolerability of GA treatment in the human patient suffering from a relapsing form of multiple sclerosis comprises reducing the frequency of an injection site reaction."
Definitions
- At [0064]-[0077] the specification defines a number of terms. The only definition which it is necessary to note is that of "tolerability" at [0066]:
"As used herein, 'tolerability' relates to the level of discomfort associated with GA treatment. Tolerability is associated with the frequency and severity of post injection reactions and injection site reactions. Tolerability influences the period that patient can follow GA treatment."
- At [0078] the specification states:
"This invention is illustrated in the Examples section which follows. This section is set forth to aid in an understanding of the invention but is not intended to, and should not be construed to, limit in any way the invention as set forth in the claims which follow thereafter."
Experimental details
- At [0079]-[0108] the specification sets out, as Example 1, a protocol for a potential Phase III clinical trial. The objective of the proposed trial is stated at [0084] to be:
"To assess the efficacy, safety and tolerability of Glatiramer Acetate (GA) injection 40mg/ml administered three times weekly compared to placebo in a double-blind study design."
- At [0096] the specification describes the dosage and route of administration for the two arms of the proposed trial, namely 40 mg GA in 1 ml for subcutaneous injection in a pre-filled syringe and matching placebo injection comprising mannitol in 1 ml water for injection also in a pre-filled syringe.
- At [0097]-[0099] the specification describes the primary and secondary outcome measures of the proposed trial, together with exploratory endpoints. The primary outcome measure is the total number of confirmed relapses during the 12 month placebo controlled phase. The secondary outcome measures are: (i) the number of new T2 lesions at month 12 as compared to baseline scan; (ii) the cumulative number of enhancing lesions on T1 weighted images taken at months 6 and 12; and (iii) brain atrophy as defined by the percent brain volume change from baseline to month 12. 12 exploratory endpoints are identified, including the time to first confirmed relapse and changes in EDSS scores and AI scores amongst others.
- The specification describes the safety and tolerability outcome measures separately at [0100]-[0101]. The tolerability measures are (i) "[p]roportion of subjects (%) who prematurely discontinued from the study, reason of discontinuation and the time to withdrawal"; and (ii) "[p]roportion of subjects (%) who prematurely discontinued from the study due to AEs and the time to withdrawal".
Results
- The "Results" section does not set out any actual results at all, but rather the desired results that the patentee hopes to achieve, set out against each primary and secondary endpoint. In relation to the primary outcome measure, these are stated at [109] as follows:
"Treatment with 40mg s.c. GA times weekly reduces the subject population annualized relapse rate by 30% or more when compared to the placebo group. Treatment with 40 mg s.c. GA three times weekly is at least effective as 20 mg s.c. GA daily administration at reducing the subject population annualized relapse rate."
- As the skilled reader would appreciate, the result set out in the second sentence cannot be demonstrated by the trial described in the specification, since the trial does not include a 20 mg arm.
- The specification does not set out desired results in relation to the tolerability measures. Nor does the patentee propose to assess the number or frequency of IPIRs or ISRs.
Discussion
- At [0112] the specification identifies the inconvenience of daily injections and the problem of ISRs as being drawbacks of GA therapy. At [0113] it states that "several obstacles and limitations with potential approaches for addressing the drawbacks exist to current GA therapy". These obstacles and limitations are all related to subcutaneous drug delivery: the acceptable volume of injection; drug degradation at the site of injection; localised trapping of the drug in the interstitial space leading to further localised irritation; and the unpredictability of the effect of variations in the frequency of administration.
- At [0114] the specification states:
"Accordingly, the subject application discloses an effective low frequency dosage regimen of GA administration to patients suffering from a relapsing form of multiple sclerosis, including patients who have experienced a first clinical episode and have MRI features consistent with multiple sclerosis. Based on the performance of the dosage regimen in these studies, the administration of three s.c. injections over a period of seven days with at least one day between every injection is also expected to work in the treatment of patients who have experienced a clinically isolated syndrome (CIS). This is based on the fact that the 20mg daily s.c. injection has been shown to work in PCT International Application PCT/US2008/03146
"
- This is an odd paragraph, because it suggests that the dosage regimen of the Patent has been shown to work in "these studies", but the specification does not report the results of any such studies. The PCT application referred to in [0114] refers to the finding that 20 mg QD and 40 mg QD doses had been shown to reduce the total number of enhancing lesions in MS patients as measured by MRI and also includes a reference to the FORTE Phase II trials (Cohen 2007). The PCT application itself relates to the efficacy for 20 mg for the treatment of CIS. Taken together, this paragraph appears to be justifying the rationale for extending the application of the claimed low frequency regimen to CIS by reference to the efficacy of the 20 mg and 40 mg QD dosages. It is not referring to any data concerning use of a 40 mg TIW regimen.
The claims
- The Patent has four claims, of which claims 1 and 3 are asserted to be independently valid.
- Claim 1 is as follows:
"Glatiramer acetate for use in a regimen of three subcutaneous injections of a 40mg dose of glatiramer acetate every seven days with at least one day between each subcutaneous injection for use in treating a patient who is suffering from a relapsing form of multiple sclerosis or who has experienced a first clinical episode and is at high risk of developing clinically definite multiple sclerosis and wherein the pharmaceutical composition further comprises mannitol."
- Claim 3 is as follows:
"Glatiramer acetate for use according to claim 1, wherein the tolerability of glatiramer acetate treatment in the human patient is increased by reducing the frequency of an immediate post injection reaction or an injection site reaction."
The skilled person
- A patent specification is addressed to those likely to have a practical interest in the subject matter of the invention, and such persons are those with practical knowledge and experience of the kind of work in which the invention is intended to be used. The addressee comes to a reading of the specification with the common general knowledge of persons skilled in the relevant art, and he or she reads it knowing that its purpose is to describe and demarcate an invention. Purely for convenience, I will hereinafter refer to the skilled person as "he". He is unimaginative and has no inventive capacity. In some cases, the patent may be addressed to a team of persons having different skills.
- There is a small, but nevertheless significant, dispute between the parties as to the identity of the skilled person or team to whom the Patent is addressed. The Claimants contend that it is addressed to a clinician who treated MS and had a research interest in the treatment of MS. Such a person would be a neurologist and would have direct experience of administrating therapeutic agents for the treatment of MS. He would have familiarity with the various dosing schedules and frequencies of the different therapeutic agents available for MS treatment. In addition, he would have had several years of experience working with the pharmaceutical industry, with experience in the design of trials necessary for drug development.
- The Defendants contend that the Patent is addressed to a person or team with the following core skills: (i) expertise in the clinical treatment of MS; (ii) expertise in the field of neurology, especially neuroimmunology (including the pathology of MS and the analysis of MRI of MS sufferers); and (iii) knowledge about clinical trial design and medical statistics.
- To the extent that there is difference between these formulations, I consider that the Claimants' formulation is to be preferred. The clinician with expertise in the treatment of MS would be a neurologist. As such, he would be expected to know about neuroimmunology, the pathology of MS and analysis of MRI in his role in diagnosing and monitoring MS patients, but only to the extent that this was relevant to his clinical practice of treating MS patients and his research in that field. The skilled person would not need to be an expert in neuroimmunology, the pathology of MS or the analysis of MRI, since the Patent does not assume any specialist knowledge of those topics or teach the clinician anything about them that he would not already know.
The expert witnesses
- The Claimants relied on expert evidence from Dr Ari Green while the Defendants relied on expert evidence from Professor Wolfgang Brück and Professor John Zajicek. Before considering the experts, I wish to say a few words about duplicative expert evidence in patent cases.
- As long ago as 1993, Aldous J stated in Gerber Garment Technology Inc v Lectra Systems Ltd [1994] FSR 471 at 489 that "[i]t is contrary to the good administration of justice that there should be duplication [of experts]". Since then the Patents Court has generally not permitted parties to call more than one expert witness in each field of expertise, and has required parties who have the same interest to share experts. Despite this, parties regularly attempt to adduce evidence from one expert which duplicates evidence given by another expert. In Regeneron Pharmaceuticals Inc v Genentech Inc [2013] EWCA Civ 93, [2013] RPC 28 Kitchin LJ (with whom Moses and Longmore LJJ agreed) stated at [125]:
"
as a general matter, it is highly undesirable for a party to adduce evidence from two different experts on the same issue. It is likely to lead to an increase in the cost and complexity of the case and to provide no corresponding benefit to the court in dealing with the case justly and in accordance with the overriding objective. Moreover, it is likely to create practical difficulties for the party faced with the evidence of precisely the kind which the appellants say arise in this case. But that party should raise the issue with the judge, preferably before or, at the latest, during the trial, and seek appropriate directions as to whether the party seeking to rely upon the evidence should be permitted to do so and, if he is, the appropriate course to be adopted in relation to it.
"
- In the present case, it was common ground between the parties at the case management conference that there was potentially a need for expert evidence in the fields of (i) clinical treatment of MS, (ii) neurology and (iii) statistical analysis of clinical trials. The Claimants considered that it was likely to be possible to adduce evidence from one expert, whereas the Defendants considered that it might be necessary to have three experts. I gave the parties permission to call up to three experts each, but I gave the Defendants the following warning:
"I think it is going to be a recipe for trouble if you have different human beings covering both the clinical aspect and the neurological aspect. If you have read my judgments over the past couple of years where you have multiple expert cases, you will see that time and time again I have commented on difficulties that are caused by having experts that overlap with each other. It really is not helpful."
- Notwithstanding this clear warning, the Defendants proceeded to serve expert reports from Prof Brück and Prof Zajicek which substantially duplicated each other. The duplication was made manifest, rather than avoided, by the fact each expert repeatedly stated that he agreed with passages in the other expert's reports. Inevitably, this led to counsel for the Claimants having to raise on the first day of the trial the question of which expert he was required to cross-examine where their evidence overlapped. Despite the fact that, on the face of their reports, Prof Brück was the Defendants' primary expert and Prof Zajicek was apparently only being relied upon due to Prof Brück's lack of clinical experience of the tolerability of GA, the Defendants elected to rely upon Prof Zajicek's evidence in preference to that of Prof Brück where they overlapped. To this end, the Defendants produced highlighted copies of Prof Brück's reports, with the highlighted passages (which extended beyond those which Prof Zajicek had previously expressly agreed with) being adopted by Prof Zajicek and omitted from Prof Brück's evidence. In addition, the Defendants called Prof Zajicek first, although I was informed that this was due to problems with his availability. Having cross-examined Prof Zajicek, counsel for the Claimants elected not to cross-examine Prof Brück on what little was left of his reports.
- Although an acceptable solution was thus arrived at, it should be clearly understood that conduct of this kind is unacceptable. Where parties adduce evidence from more than one expert, the evidence of the respective experts should be clearly delineated. Parties who flout the guidance which the courts have repeatedly given can expect to be heavily sanctioned in costs. This may extend to their legal advisors if the circumstances warrant such an order.
- Dr Green is the Medical Director of the University of California, San Francisco ("UCSF") Multiple Sclerosis Center. He received a Bachelor of Arts and Science from Miami University in 1994, an MD from Duke University School of Medicine in 2001 and a Master's degree in Clinical Research from UCSF in 2007. He completed postgraduate medical training at UCSF, completing an internship in the Department of Medicine in 2002, and a residency in Neurology in 2004. He became the Chief Resident of Neurology from 2004-2005, and a Clinical Fellow in the Departments of Neuro-Ophthalmology and Neuro-Immunology from 2005 to 2007. In 2006 he was a Visiting Fellow in the Department of Neuropathology (Retina) at the Queen's University in Belfast, where he observed clinical treatment of patients with MS in the UK. He was a Clinical Instructor in the Department of Neurology from 2005-2007, an Assistant Professor of Clinical Neurology from 2007 to 2013 and an Associate Professor of Neurology and Associate Professor of Ophthalmology since 2013. He has served as the Medical Director of the Multiple Sclerosis Center since 2012. He has been treating MS patients since the start of his medical career, prescribing GA 20 mg QD from 2002 onwards. He has published nearly 70 papers and has been involved in 16 clinical trials in one capacity or another. Dr Green also testified as an expert witness in proceedings concerning US equivalents of the Patent before the District Court of Delaware and before the Patent Trial and Appeal Board of the US Patent and Trademark Office, as well as in two other cases concerning GA.
- Counsel for the Defendants submitted that Dr Green had become entrenched in his views as a result of his prior experience as an expert witness. I do not accept this. Although Dr Green's oral evidence was generally consistent with his written reports, he did make certain concessions in cross-examination. Counsel for the Defendants also made three specific criticisms of Dr Green's evidence. The first was that Dr Green had applied a different standard to Devonshire and to the GLACIER trial (as to both of which, see below). I do not accept this: Dr Green's evidence was that there were key methodological flaws in the GLACIER trial, whereas it was not established that Devonshire suffered from similar flaws, although Dr Green did accept that the study had statistical limitations. The second criticism was that, in his discussion of Devonshire in his first report, he presented a graph which showed a trend of increasing non-adherence with increased frequency of administration of DMTs, but did not include a data point for 22 µg Rebif in addition to the data point for 44 µg Rebif. I accept Dr Green's explanation that he did so for simplicity, given that the 44 µg dose had a higher rate of non-adherence than the 22 µg dose and that there is no indication that the difference in rates between the 44 µg and 22 µg doses was statistically significant. The third criticism concerned his failure to mention in his reports that there appeared to have been a problem with randomisation in slide 22 of Comi 2008B (as to which, see below) even though he had spotted this while being deposed in the US. I agree that this is a point that Dr Green should have mentioned in his reports, but he readily acknowledged it in cross-examination, and I regard this as an isolated lapse of no greater significance. As counsel for the Claimants pointed out, Prof Brück only picked up this point in his third report despite that the fact that his first report for the EPO proceedings was filed in October 2015; prior to his third report, Prof Brück's evidence on this aspect of Comi 2008B had been in accordance with Dr Green's. Overall, I found Dr Green to be an excellent witness whose answers were clear, balanced and well-reasoned.
- Prof Zajicek is Chair of Medicine at the University of St Andrews and Honorary Consultant neurologist for NHS Fife. He obtained a Medical Sciences Tripos degree from the University of Cambridge in 1981, followed by MB.BS (Hons) from the University of London in 1984, before going on to hold various pre-registration house roles, senior house officer roles and registrar roles at a number of different hospitals around the country, first specialising in neurology in 1987. He began to specialise in the treatment of MS in 1989 when he was appointed Registrar in Neurology at Addenbrooke's Hospital, Cambridge. In 1990 he became a Wellcome research fellow at the University of Cambridge and was awarded a PhD in 1994 for research into the mechanisms of MS. In 1995 he became a Consultant Neurologist at the Plymouth Hospitals NHS Trust. In 2000 he was appointed a Senior Lecturer in Neurology at the University of Plymouth, and then Reader in 2001. He was appointed Professor in Clinical Neuroscience and Professor of Medicine at the University of St Andrews in 2015. He has published approximately 100 papers relating to MS, with much of his research being devoted to the effects of cannabis and cannabinoids on the symptoms of MS. Since 1995 he has been the Principal Investigator in approximately 40 pharmaceutical industry-sponsored clinical trials. He has also been co-investigator on trials with various companies including trials involving DMTs (GA, interferons and fingolimod).
- Counsel for the Claimants submitted that Prof Zajicek had been put in a difficult position by the Defendants through being asked to adopt passages in Prof Brück's reports which were inconsistent with Prof Zajicek's evidence as to the weight which the skilled person would place on prior publications concerning lower frequency administration of GA. Counsel for the Claimants accepted that Prof Zajicek's oral evidence was generally fair, although he criticised some of the answers Prof Zajicek gave as not being credible or consistent. These submissions accord with my assessment of Prof Zajicek's evidence.
- Counsel for the Claimants also submitted that Prof Zajicek had adopted an approach to the various publications concerning trials of GA prior to August 2009 which did not correspond with the approach which the evidence showed had actually been adopted by workers in the field at the time, and hence would not have been adopted by the skilled person to whom the Patent was addressed. In his reports, Prof Zajicek expressed the opinion that the skilled person would not "place any weight" on the results of trials which had not been properly conducted (a properly conducted trial being one that complied with the standards laid down in the CONSORT Statement: see Moher et al, "The CONSORT statement: revised recommendations for improving the quality of reports of parallel-group randomised trials", Lancet, 357, 1191-94 (2001)). Moreover, he adopted passages of Prof Brück's reports expressing the opinion that the skilled person would not draw "useful" or "meaningful" conclusions from such results. As counsel for the Defendants pointed out, Prof Zajicek somewhat modified his stance on this point during the course of cross-examination, when he agreed that clinical trials which had not been properly conducted could nevertheless yield scientifically valuable data. Even so, I consider that the evidence shows that workers in the field placed more weight upon less-than-scientifically-rigorous trials and analyses of results than Prof Zajicek was prepared to accept even in cross-examination. A clear example of this, although there are others, is provided by the FORTE Phase II trial reported in Cohen 2007: Prof Zajicek's evidence was that the skilled person would not have considered that the results warranted proceeding to a Phase III trial, whereas what actually happened was that the Phase III trial was proceeded with. It is immaterial that the results of the Phase III trial show that Prof Zajicek's reservations were scientifically justified.
- Prof Brück is Professor of Neuropathology and Head of the Department of Neuropathology at the University Medical Center at Georg August University Göttingen in Germany. He obtained his medical degree from the Johannes Gutenberg University in Mainz, Germany in 1986 and then took up a residency in Neuropathology at Georg August University Göttingen from which he obtained his "habilitation" (a professorial qualification) in Neuropathology in 1996. From 1995 to 1999, he was also a Consultant in neuropathology in the Department of Neuropathology at Georg August University. From 1999 to 2002 he held an Associate Professorship at the Humboldt University in Berlin in the Department of Neuropathology at the Charité (the hospital of Humboldt University in Berlin), before returning to Göttingen as Professor and Head of the Department of Neuropathology. Since the early-1990s the major focus of his work has been MS, and in particular the pathology and immunopathology of MS. His work encompasses both the clinical aspects of MS (in particular assisting with the diagnosis of the disease) and research into the pathology and pathogenesis of MS. He has been involved in clinical trials in an advisory capacity. Prof Brück also gave evidence in European Patent Office opposition proceedings concerning the parent of the Patent and in parallel Dutch proceedings.
- Counsel for the Claimants submitted that what remained of Prof Brück's evidence following the exercise described in paragraph 82 above was either irrelevant or of no assistance to the court. It was irrelevant because Prof Brück gave evidence in his reports from the perspective of a person with expertise in the pathology and neurology of MS and analysis of MRI, and not from the perspective of a person with experience of the clinical treatment of MS. In particular, he had no experience of the tolerability issues experienced by patients, which was why the Defendants had adduced evidence from Prof Zajicek. It was of no assistance because Prof Brück gave no evidence about the Patent, the prior art, obviousness or plausibility. Nor did Prof Brück (or Prof Zajicek) give any evidence explaining how Prof Brück's skilled person would have interacted with Prof Zajicek's skilled person in a skilled team.
- I accept these submissions, which are reinforced by two points. The first is that, when I asked counsel for the Defendants what expertise Prof Zajicek had lacked, he was unable to identify any. The second is that both Prof Brück and Prof Zajicek gave evidence that the first step that their respective skilled persons would have taken would have been to carry out a literature search, yet the searches they undertook were different and produced different results. In particular, Prof Brück's search included publications relating to in vitro studies and animal models, whereas Prof Zajicek excluded such publications from his search on the ground they were not predictive of efficacy in humans, particularly with respect to dosing regimens. As counsel for the Claimants submitted, it is clear from Prof Zajicek's evidence that the skilled person to whom the Patent is addressed would, if he had performed a literature search, have carried out the one performed by Prof Zajicek.
- Finally, I would add that the editing process resulted in the remaining parts of Prof Brück's evidence being rather disjointed.
- The upshot of these considerations is that I conclude that, where there is a conflict between the evidence of Dr Green and that of Prof Zajicek and/or Prof Brück, the evidence of Dr Green carries more weight. It remains necessary, however, for me to consider the cogency of the experts' reasons for the opinions they expressed.
Common general knowledge
- I reviewed the law as to common general knowledge in KCI Licensing Inc v Smith & Nephew plc [2010] EWHC 1487 (Pat), [2010] FSR 31 at [105]-[115]. That statement of the law was approved by the Court of Appeal [2010] EWCA Civ 1260, [2011] FSR 8 at [6].
- The Claimants accept that this Court should proceed on the basis that it must be shown that information was common general knowledge in the UK and that it is not sufficient to show that it is common general knowledge elsewhere in the world, such as in the USA (see Generics (UK) Ltd v Warner-Lambert Co LLC [2015] EWHC 2548 (Pat), [2016] RPC 3 at [123]-[124]), but reserve the right to contend to the contrary in a higher court.
- It is common ground that everything I have set out in the technical background section of this judgment formed part of the skilled person's common general knowledge. There are a number of topics as to which there is some disagreement as to the extent of the common general knowledge, however. My findings are set out below.
- It is also common ground that, in some cases, it may be obvious for the skilled person to undertake a literature search: see Generics (UK) Ltd v Daiichi Pharmaceutical Co Ltd [2009] EWCA Civ 646, [2009] RPC 23 at [26]-[27]. In such a case, while the information which would be ascertained by means of the literature search may not be common general knowledge, it is legitimate to take it into account when considering obviousness. In the present case, it is convenient to consider this question together with that of common general knowledge.
US versus UK knowledge
- It is common ground that the evidence shows that MS and its possible treatments were widely discussed internationally, that neurologists from all over the world attended international conferences and read the same leading journals and that many clinical trials involved international collaborations. Accordingly, it is common ground that in most respects the common general knowledge of the skilled person in the UK was the same as that of the skilled person elsewhere in the world, and in particular the USA.
- The Defendants nevertheless contend that there were significant differences in the way MS was treated in the UK and in the US, which would give rise to differences in clinical experience. I accept that the evidence shows that there were some differences. The most relevant difference is that, in the UK, patients were extensively counselled prior to starting treatment with DMTs, which led to greater adherence rates than in the US.
Side effects of the DMTs
- Prof Zajicek's evidence was that, as at August 2009, it was thought that the side effects of GA were generally less severe than those of the interferons. GA did not cause the flu-like symptoms that the interferons did, and it led to fewer ISRs. Dr Green agreed that that had been the position earlier in the decade, but disagreed that it was still the case in August 2009. He relied upon the BEYOND (Betaferon Efficacy Yielding Outcomes of a New Dose) and PreCISe trials, the results of which were presented at the 60th annual meeting of the American Academy of Neurology in Chicago in April 2008 and widely reported (in particular, in an article by Susan Jeffrey in Medscape dated 17 April 2008). The trials were not fully reported until after August 2009, however (in the case of BEYOND, in O'Connor et al, "250 µg or 500 µg interferon beta-1b versus 20 mg glatiramer acetate in relapsing-remitting multiple scleroses: a prospective, randomised, multicentre study", Lancet Neurol., 8, 889-897 (October 2009), and in the case of PreCISe, in Comi et al, "Effect of glatiramer acetate on conversion to clinically definite multiple sclerosis in patients with clinically isolated syndrome (PreCISe study): a randomised, double-blind, placebo-controlled trial", Lancet, 374, 9700 (31 October 2009)). It was not put to Prof Zajicek that the results of the BEYOND and PreCISe trials were common general knowledge in August 2009. Accordingly, I am not satisfied that this was established.
- On the other hand, the skilled person would have known that the Copaxone 20 mg QD Summary of Product Characteristics stated:
"In all clinical trials, injection-site reactions were seen to be the most frequent adverse reactions and were reported by the majority of patients receiving Copaxone. In controlled studies, the proportion of patients reporting these reactions, at least once, was higher following treatment with Copaxone (70%) than placebo injections (37%)."
Similarly, the Patient Information Leaflet stated that ISRs were "very common (more than 1 in 10 patients)".
Adherence and convenience
- It is common ground that DMTs differ from other drugs used to treat RRMS in that they are a form of prophylactic therapy, designed to reduce the number of relapses suffered by a patient and hopefully prevent long-term progression of the disease. As such, the patient does not notice an immediate benefit of taking DMTs. This is in contrast with, for example, the patient experience on corticosteroids, which are designed to accelerate recovery from acute relapses, and other forms of therapy which are designed to treat existing symptoms such as fatigue, pain, muscle spasticity and visual defects.
- There is disagreement as to the extent to which the skilled person would have regarded patient adherence (the extent to which a patient's behaviour corresponds with a caregiver's instructions, sometimes referred to as patient compliance, at least in the context of whether medication is taken as prescribed), as a significant issue with DMTs in August 2009. Dr Green's evidence was that his experience was that patient non-adherence was up to 50% for injectable immunomodulatory therapies in MS and that the degree of patient non-adherence increased as the frequency of injections increased. Dr Green explained that there were two related reasons for this: first, the more frequent the injections, the more likely it was that an ISR would be experienced; secondly, the more painful the injection, the more likely the patient would be to miss a dose.
- Although Dr Green said that his experience was reflected in several studies, he only made specific reference to one, namely Devonshire et al, "The Global Adherence Project a multicenter observational study on adherence to disease-modifying therapies in patients suffering from relapsing-remitting multiple sclerosis", Multiple Sclerosis 12, S82 (2006) ("Devonshire"). This was a survey of 2646 patients asking them to report non-adherence (defined as missing at least one DMT injection or changing dose within four weeks prior to the survey). The authors found that the non-adherence rate went up as the frequency of injections went up: 15% for Avonex (a once weekly administration), 22.0% for Rebif 22 µg (TIW), 27.3 % for Rebif 40 µg (TIW), 30.9% for Betaseron (QOD) and 34.2% for Copaxone (QD). They also found that the difference between the non-adherence rate for Avonex and the non-adherence rates for the other DMTs was statistically significant.
- The Defendants advance a number of criticisms of Dr Green's reliance upon Devonshire. First, they contend that Devonshire shows that non-adherence rates are not solely dependent on frequency of injection, but also on dose. There is nothing to show whether the difference between Rebif 22 µg and Rebif 44 µg was statistically significant or not, however. Secondly, and inconsistently with their first point, they point out that it does not show whether or not the non-adherence rates for Rebif 40 µg, Betaseron and Copaxone were statistically different from each other. Thirdly, they point out that frequency of administration is not the only difference between the regimens of the various DMTs. Fourthly, they point out that the figures were not adjusted for the number of drug days missed.
- In relation to the last point, the Defendants rely upon a study which Prof Zajicek exhibited to his second report (Treadway et al, "Factors that influence adherence with disease-modifying therapy in MA", J. Neurol., 256, 568-576 (2009), "Treadway"), which found that only Betaseron had a significantly different adherence rate from the others on a drug-days-missed basis. Dr Green's view was that that was not a particularly informative analysis, however. Moreover, he pointed out that both Devonshire and Treadway found (consistently with his own clinical experience) that the most common reason for non-adherence was that the patient forgot to administer an injection and that that was a bigger problem with drugs that were more frequently administered.
- Turning to Prof Zajicek, he explained that in his experience problems with patient adherence could be reduced, if not eliminated, by carefully counselling the patients before they started treatment. Nevertheless, he accepted that it was well known to the skilled person that ISRs from injections probably caused a reduction in adherence, and therefore negatively affected patients' use of all the available drugs, although his view was that ISRs were not much of problem for GA. He also accepted that reducing the frequency of injections of GA at a given dose would be expected to reduce the frequency of ISRs and IPIRs. On the other hand, he pointed out that some patients found it easier to remember to take their medication with daily administration.
- In addition to Devonshire and Treadway, a number of other publications in evidence speak to the issue of adherence including Filippi 2006 and Khan 2008 (as to which, see below). Thus the authors of Filippi 2006 state in the introduction at 213:
"All currently approved drugs for multiple sclerosis are administered parenterally. However, long-term treatment with injected drugs is not without problems. These include patient discomfort and the occurrence of adverse events associated with frequent injections, such as local injection site reactions. These issues, through reduction of patient compliance, probably negatively affect patients' use of all available drugs. Thus, there is a strong rationale for assessment of whether drugs that are known to be effective when given parenterally also exert positive effects on clinical and MRI measures of disease activity when given orally."
It was on this basis that the CORAL Phase III trial was undertaken.
- The conclusion I reach from the evidence as a whole is that the skilled person would have known that patient adherence was a problem with DMTs, including GA, although it could be reduced by careful counselling. The skilled person would also have known that reducing the frequency of injections of GA at a given dose would be expected to improve patient adherence for two reasons: first, because it would reduce the frequency of ISRs and IPIRs, and secondly, because it would tend to reduce the number of injections that were missed through forgetfulness.
- Patient convenience is a distinct, although related, issue to that of patient adherence. Dr Green and Prof Zajicek were agreed that less frequent injections were more convenient for patients. As Prof Zajicek explained in his first report:
"In my experience, for most of my patients the frequency of injections was an important factor in selecting which treatment to take. For [patients with mild RRMS] Avonex was a popular choice because it was administered once weekly (albeit via intramuscular injection which can be more painful than sc injections) and led to fewer ISRs than the more frequently administered interferons."
- Dr Green and Prof Zajicek were also agreed that QOD regimens had the disadvantage for patients that they had to remember on which days to inject, which varied from week to week, whereas a TIW regimen permitted dosing on the same days each week. A TIW regimen would also permit the patient to have weekends injection-free, which some patients would regard as advantageous.
The 20 mg QD regimen for GA administration
- It was common ground between Dr Green and Prof Zajicek that the skilled person would have been aware that the 20 mg QD regimen for GA appeared to have been somewhat arbitrarily selected and that no Phase II dose-ranging studies had been carried to determine the optimum dose of GA for treating MS, and in particular RRMS. It was Dr Green's evidence that, despite the uncertainty over GA's mechanism of action, the skilled person would have expected its effects to last longer than 24 hours. This was not put to Prof Zajicek, however, and so I am not satisfied that this was shown to be common general knowledge.
The FORTE trials
- Although there was some disagreement as to the extent to which the detailed results of the FORTE trials were common general knowledge in August 2009, it is common ground that this does not matter, because it would be obvious for the skilled person who was considering a new dosing regimen for GA to carry out a literature search which would turn up Cohen 2007 and Comi 2008A and to obtain Comi 2008B. What does matter is that there is considerable disagreement as to what the skilled person would have taken from those publications.
- Cohen 2007 is summarised in the abstract as follows:
"Objective: To evaluate the safety, tolerability, and efficacy of glatiramer acetate (GA) 40 mg daily vs. the approved 20 mg formulation in relapsing remitting multiple sclerosis. Methods: Eligibility criteria included clinically definite multiple sclerosis, expanded Disability Status Scale score 0 to 5.0, no previous use of GA, at least one relapse in the previous year, and 1 to 15 gadolinium-enhancing (GdE) lesions on a screening MRI. MRI was repeated at months 3, 7, 8, and 9, and neuralgic examinations were performed at baseline and months 3, 6, and 9. Results: Of 229 subjects screened, 90 were randomly assigned to GA 20 mg (n = 44) or 40 mg (n= 46). The groups were well matched at baseline for demographic, clinical, and MRI characteristics. The primary efficacy endpoint, total number of GdE lesions at months 7, 8 and 9, showed a trend favouring the 40-mg group (38% relative reduction, p = 0.0898). A difference between the two dose groups emerged as early as month 3 (52% reduction; p = 0.0051). There was a trend favouring the 40-mg group for relapse rate with benefit on proportion of relapse-free subjects (p = 0.0183) and time to first relapse (p = 0.0367). GA 40 mg was well tolerated, with an overall safety profile similar to that of 20 mg. Some features of injection site reactions and immediate postinjection reactions were more common and severe with the higher dose. Conclusions: Glatiramer acetate (GA) 40mg was safe and well tolerated. The overall efficacy results suggested that a 40-mg dose of GA may be more effective that the currently approved 20-mg dose in reducing MRI activity and clinical relapses."
- The MRI results are shown graphically in figure 2, which I reproduce below:
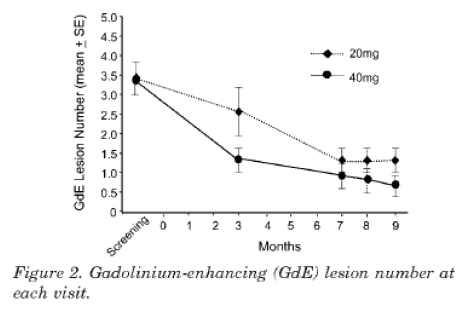
- In the Results section of the article, the authors report the following findings with respect to tolerability at 942-943 (footnotes omitted):
"Injection site reactions were the most frequent adverse event for both doses, occurring in 38 (86.4%) subjects in the 20-mg group and 39 (84.8%) in the 40-mg group. Injection site manifestations reported with at least 5% higher incidence for GA 40 mg included burning, mass, pass, and urticaria (table 3). Skin necrosis and lipoatrophy were not observed. Thirty-nine IPIRs occurred in 10 (22.7%) subjects on GA 30 mg vs 52 IPIRs in 15 (32.6%) subjects on GA 40 mg. The incidence in the 200mg group was consistent with previous large studies of GA. IPIRs were categorized most often as moderate severity in the 40-mg group and mild in the 20-mg group. All IPIRs resolved without sequelae, although one in the 40-mg group led to hospitalization and subsequent discontinuation from the study. The greatest difference in IPIR component symptoms
was in palpitations."
- In the Discussion section of the article, the authors comment on the MRI results shown in figure 2 and on the tolerability findings at 944 as follows (footnote omitted):
"In the present study, an advantage of the 40 mg dose on suppressing GdE lesions was already evident at month 3 and, in fact was more prominent compared with months 7, 8, and 9. Similarly, results based on the first relapse, e.g. time to first relapse and proportion of relapse free subjects, were more robust than measure based on the 9 month study period, e.g., relapse rate. These results suggest that the onset of action of the 40 mg dose is more rapid compared with 20 mg. A larger, longer study will be necessary to confirm the sustainability of the efficacy advantage of the higher dose.
GA at the currently approved 20 mg dose has been safe and well tolerated in previous trials and post-marketing experience. The overall safety and side effect profile of the 40 mg dose in this trial was similar, although it was associated with greater incidence of certain adverse effects. For example, although the overall incidence of injection site reactions was similar, some aspects were more common with the higher dose, and the injections seemed to be somewhat more painful. IPIRs also were somewhat more common and severe. Qualitatively, both side effects were similar what has been seen previously."
- Comi 2008A is sufficiently brief for me to set it out in full:
"Background: Results from a phase II study showed that 40mg dose of glatiramer acetate (GA) was possibly more effective than the currently approved 20mg dose in reducing MRI activity and clinical relapses. Objective: To evaluate the safety, tolerability, and efficacy of GA 40mg compared to the 20mg dose. Methods: Patients with definite MS (revised McDonald criteria), with at least one documented relapse in the 12 months prior to screening, or at least two documented relapses in the 24 months prior to screening, and EDSS score 05.5, were enrolled. Subjects were evaluated at screening, baseline, and at months 1, 2, 3, 6, 9, and 12. The primary endpoint was the rate of confirmed relapses observed during the study. Results: A total of 136 centers in 20 countries participated in the trial. Of the 1262 subjects screened, 1155 were randomized to GA 20mg (n=586) or 40mg (n=569). The groups were well-matched at baseline on demographic, clinical, and MRI characteristics. Annualized relapse rate (ARR) in the last year prior to study was 1.498. The primary efficacy endpoint was similar in both groups (relative risk=1.07, p=0.4859) with mean ARR of 0.33 for the 20 mg and 0.35 for the 40 mg group and 0.27 for those who completed one year treatment. Seventy-seven % of the patients remained relapse-free in both groups. Both groups showed a reduction in the mean number of gadolinium-enhancing and new T2 lesions over time with a trend for a faster reduction in the first trimester in the 40mg dose compared with 20mg dose. Both doses were well-tolerated with a safety profile similar to that observed in previous studies of 20mg GA. Conclusions: In RRMS patients, both GA 40 mg and the currently approved 20mg doses were safe and well-tolerated, and were equally effective in reducing clinical relapses and MRI activity."
- Comi 2008B contains more detail about the trial and the results. Slide 5 headed "Background" states:
"* Three pivotal trials support the efficacy, tolerability, and safety of GA 20 mg dosing in RRMS patients
* Results from a phase II study showed that 40mg dose of glatiramer acetate (GA) was possibly more effective than the currently approved 20mg dose in reducing MRI activity and clinical relapses".
- I reproduce slide 14 below:
- It can be seen from the first footnote that the increase in the percentage of early terminations due to adverse events from 4.8% with 20 mg to 9.0% with 40 mg is said to be statistically significant and to be mainly due to ISRs.
- I reproduce slide 22 below:
- The slide draws attention to a 38% difference between the 40 mg group and the 20 mg at three months, suggesting a faster onset of action with the 40 mg dose. It can be seen, however, that the 40 mg group started with a higher number of lesions than the 20 mg group, and thus there appears to have been a problem with the randomisation in that respect. It can also be seen that the two groups had the same number of lesions at 12 months.
- Slide 25 sets out of the following data for safety outcomes: serious adverse events 4.3% for 20 mg and 4.2% for 40 mg, ISRs 55.6% for 20 mg and 58% for 40 mg and IPIRs 6.1% for 20 mg and 7.6% for 40 mg. There is no statement that the differences are statistically significant. The slide then states:
"* Safety profile similar to that observed in previous studies of GA 20mg; both doses were well-tolerated; increased treatment discontinuation due to injection site reactions with the higher dose
* No safety concerns in both doses with regards to Laboratory Results, ECG and Vital signs".
- Slide 26 headed "Summary and Conclusions" states:
"* No significant differences in efficacy measures between GA 20mg and GA 40mg
* Both doses demonstrated remarkable reduction compared to baseline in clinical and MRI activity
* Good safety and tolerability profile; no unexpected adverse effect with the higher dose
* Trend for an earlier effect of high dose on MRI activity".
- It does not appear that either Dr Green or Prof Zajicek attended Prof Comi's presentation, but Prof Brück did. Subsequently Prof Brück briefly summarised the results of the FORTE Phase III trial in two slides of a presentation to the MS Quality Circle at his hospital in Göttingen on 2 February 2009. Counsel for the Defendants relied upon these slides as contemporaneous evidence uninfluenced by the exigencies of litigation of what a skilled person would take away from the presentation. Even if it is assumed, contrary to the conclusion I have reached above, that Prof Brück was representative of the skilled person to whom the Patent is addressed, however, as counsel for the Defendants himself submitted, the skilled person would have read and considered Comi 2008B as a whole.
- Cohen 2007, Comi 2008A and Comi 2008B were considered at some length in the expert evidence and in the parties' closing submissions. Considering the evidence of the experts as a whole in the light of the parties' submissions, I conclude that what the skilled person would have derived from these publications was as follows:
i) There was no statistically significant difference in efficacy between the 40 mg dose and the 20 mg dose in terms of MRI activity or reduction in relapses.
ii) There was some evidence in both FORTE Phase II and Phase III of an earlier onset of activity as measured by MRI with the 40 mg dose than with the 20 mg dose; but the evidence was weak, in particular because of the problem with the randomisation of the frequent MRI cohort in Phase III which meant that the apparent difference at three months could be an artefact. Moreover, there was no discussion of the clinical significance of an earlier onset of action if there was one. Dr Green and Prof Zajicek were disagreed as to whether the skilled person would consider that an earlier onset of action would be clinically beneficial, and I conclude that the skilled person would regard this as uncertain.
iii) In terms of efficacy, therefore, there was no disadvantage in administering a 40 mg dose rather than a 20 mg dose, and there was a possible, but unproven, short-term advantage of uncertain clinical significance.
iv) In the Phase II trial, there had been no real difference in the rate of ISRs between the 40 mg dose and the 20 mg dose, although there was a suggestion that injections were more painful; whereas there was an increase in both the rate and the severity of IPIRs, although it is not clear whether this was statistically significant. In the Phase III trial, there had been a statistically significant increase in early terminations due to adverse events, which were mainly due to ISRs. So far as one can tell from Comi 2008B, this was due to an increase in the frequency of ISRs, rather than their severity. There was also an increase in the frequency of IPIRs, although it is not clear whether this was statistically significant. There is no mention in Comi 2008B of the IPIRs having been more severe.
v) Nevertheless, both the 40 mg and the 20 mg doses were safe and well tolerated.
vi) In terms of tolerability, therefore, there was a disadvantage in administering a 40 mg dose rather than a 20 mg dose, mainly in terms of the frequency of ISRs, albeit that it remained well tolerated.
Work on lower frequency administration of GA
- It is common ground that the skilled person would be aware from his common general knowledge that Professor Omar Khan (Director of the Multiple Sclerosis Center and Image Analysis Laboratory at Wayne-State University School of Medicine, Detroit and a well-known figure in the MS field) and his co-workers were investigating less frequent administration of 20 mg GA. It is also common ground that the skilled person would either be aware of, or find as a result of his literature search, a total of three publications on studies involving the administration of 20 mg QOD, namely:
i) Flechter et al, "Comparison of glatiramer acetate (Copaxone®) and interferon ß-1b (Betaferon®) in multiple sclerosis patients: an open-label 2-year follow-up", J. Neurol. Sci., 197, 51-55 (2002) ("Flechter 2002A");
ii) Flechter et al, "Copolymer 1 (Glatiramer Acetate) in Relapsing Forms of Multiple Sclerosis: Open Multicenter Study of Alternate-Day Administration", Clin. Neuropharmacol., 25, 11-15 (2002) ("Flechter 2002B"); and
iii) Khan et al, "Randomized, prospective, rater-blinded, four-year pilot study to compare the effect of daily versus every-other-day glatiramer acetate 20mg subcutaneous injections in relapsing-remitting multiple sclerosis", Multiple Sclerosis, 14, S296 (2008) ("Khan 2008"). Khan 2008 is the abstract from a presentation by Prof Khan at WCTRIMS in September 2008.
- Flechter 2002A reported an open-label study involving 58 patients with relapsing forms of MS who received either (a) 20 mg GA QD or (b) 20 mg GA QOD or (c) interferon beta-1b QOD. It was found that there was a significant benefit of decreasing relapse rate in all three groups, with no significant difference between them. The authors concluded at 55 (footnotes omitted):
"In summary, although our study is an open-label one, it shows that IFNß-1b injected subcutaneously on alternate day, and Copaxone injected subcutaneously either daily or on alternate day, may modulate disease activity in a similar way and to a similar extent.
The choice of the appropriate medication for each patient still remains an individual decision since the effects of IFNß-1b and Copaxone seem to be similar. The fact that Copaxone injected on alternate day seems to be equally effective as the Copaxone injected daily is important to patients with injection-related adverse events and, from an economic point of view, as a cost-effective measure. Moreover, considering that there is no available data supporting daily vs. alternate-day Copaxone injection, in our view a well-designed post-marketing study is required, in order to confirm or refute the results obtained in our study."
- Flechter 2002B reported an open-label study involving 68 patients with relapsing forms of MS who received 20 mg GA QOD. It was found that administration QOD was efficacious, and that the efficacy was comparable to that reported in studies of QD administration. The authors concluded at 15:
"The results of this trial suggest that alternate-day treatment with Copolymer 1 [i.e. GA] is safe, well tolerated, and probably as effective as daily Copolymer 1 in reducing relapse rate and slowing neurologic deterioration. However, these preliminary observations will have to be examined in larger studies, preferably comparing daily with alternate-day administration of Copolymer 1 in a blinded manner."
- Khan 2008 is sufficiently brief to set out in full:
"Background: The recommended dose of GA in RRMS is 20 mg subcutaneous (SC) daily (QD) although the optimal dose remains unknown. There is considerable interest in alternate dosing regimens of GA in RRMS. Daily SC injectable therapy can be challenging for long-term patient compliance. Objective: We conducted a pilot trial to compare the effect of GA 20 mg SC daily versus every other day (QOD) in RRMS. The primary endpoint was based on a composite of clinical, MRI, and immunologic outcomes. Methods: Treatment naïve RRMS patients were randomized to GA 20 mg SC QD or QOD and followed prospectively for 2 years. After 2 years, patients in each group were given the option to continue or switch to the other group, and followed for an additional 2 years. EDSS was recorded every 6 months by a rater blinded to dosing allocation. Brain MRI scans were obtained at baseline, and years 2 and 4. Blood for immunologic testing was obtained at baseline and multiple time points after randomization. Results: 30 patients were randomized to GA 20 mg SC given QD or QOD. Both groups were well-matched for age, disease duration, EDSS, relapse rate, T2W and gadolinium (Gd) enhancing lesions. After 2 years, there were no differences in the relapse rate, disease progression, Change in T2W lesion volume, or Gd enhancing lesions between the two groups. In vitro proliferation of GA-responsive T-cells and Th1/Th2 cytokine expression did not differ between the two groups at any time point after randomization. After 2 years, all patients in the QD group opted to switch to QOD. After a total of 4 years of prospective follow-up, there was no difference between the QD-QOD cross over group and the always QOD group. Additional data on imaging and immunologic outcomes will be presented. Conclusions: This pilot study suggests that GA 20 mg SC administered QD or QOD may be equally effective in RRMS. This may have implications for the long-term use of GA. However, large multi-center studies are warranted to confirm our findings and to identify the optimal dose of GA in RRMS."
- Again, there is disagreement as to what the skilled person would take from these three publications. Again, they were considered at some length in the expert evidence and in the parties' closing submissions. Considering the evidence of the experts as a whole in the light of the parties' submissions, I conclude that what the skilled person would have derived from these publications was as follows:
i) There was considerable interest in less frequent administration of GA, in particular because of the adherence issue with QD administration.
ii) The studies in Flechter 2002A and Flechter 2002B suffered from methodological flaws which reduced the weight that could be placed on them, but would not lead the skilled person to disregard them altogether.
iii) Each of the studies suggested that 20 mg QOD might be as effective as 20 mg QD, but given their small scale and open-label (i.e. unblinded) nature, no firm conclusions could be drawn and larger, blinded studies were required to confirm the findings.
New treatments on the horizon
- It is common ground that in August 2009 the skilled person would have been aware that there were new treatments for RRMS on the horizon, including fingolimod, dimethyl fumarate and alemtuzumab, which were then in clinical trials. Both fingolimod and dimethyl fumarate were orally administered. There was considerable excitement about these potential new treatments, and therefore these were much more discussed than improvements in GA therapy.
Claim interpretation
The law
- Prior to 12 July 2017 it was settled law that patent claims were to be interpreted in accordance with the principles established by the House of Lords in Catnic Components Ltd v Hill & Smith Ltd [1982] RPC 183 and Kirin-Amgen Inc v Hoechst Marion Roussel Ltd [2004] UKHL 46, [2005] RPC 9 and a multitude of decisions of the Court of Appeal applying those principles. In the briefest summary, the ultimate question was what the person skilled in the art would have understood the patentee to have used the language of the claim to mean. This involved giving the claim a purposive construction, that is to say, interpreting it having regard to the fact that the patentee's purpose was to describe and claim an invention.
- In Actavis UK Ltd v Eli Lilly and Co [2017] UKSC 48 the Supreme Court departed from Catnic and Kirin-Amgen (albeit without expressly invoking the Practice Statement (Judicial Precedent) [1964] 1 WLR 1234, which "has as much effect in [the Supreme Court] as it did before the Appellate Committee in the House of Lords", see Austin v Southwark London Borough Council [2010] UKSC 28, [2011] 1 AC 355 at [25] (Lord Hope of Craighead)) at least with respect to the correct approach to equivalents to elements specified in the claims for the purposes of determining the scope of protection of patents in the context of an issue as to infringement. One of the questions which arises in the light of the Supreme Court's decision is whether, before one comes to any question of equivalents, it remains the law that patent claims should be given a purposive construction.
- Counsel for the Defendants submitted that this was no longer the law, and that instead a patent claim should be interpreted literally by which counsel explained that he meant in the same manner as a clause in a commercial contract and without regard to the patentee's purpose. In support of this submission, he relied upon the following paragraphs of the judgment of Lord Neuberger (with whom the other members of the panel agreed): [54], where Lord Neuberger referred to "normal interpretation" of claims; [58], where he said that "the applicable principles" for "interpreting documents" were those summarised by Lord Hodge in Wood v Capita Insurance Services Ltd [2017] UKSC 24, [2017] 2 WLR 109 at [8]-[15]; and [66(i)], where he referred to "the literal meaning of the relevant claim(s)".
- Counsel for the Claimants submitted that it remained the law that a patent claim should be given a purposive construction. In support of this submission, he relied upon the following paragraphs of Lord Neuberger's judgment: [22], where he said that "a patent is to be interpreted on the basis that it is addressed to a person or group of persons who is or are likely to have a practical interest in the claimed invention"; [54], where he said that both the issues of (i) "normal interpretation" and (ii) infringement by immaterial variants should be "considered through the eyes of a notional addressee" of the patent; and [56], where he referred to issue (ii) involving "not merely identifying what the words of a claim would mean in their context to the notional addressee".
- In my judgment counsel for the Claimants is correct. As has often been pointed out, patents differ from commercial contracts in two key ways. First, a contract is (at least in principle) a bilateral statement agreed between the contracting parties, whereas a patent is a unilateral statement made by the patentee and addressed to the class of persons represented by the person skilled in the art. Secondly, whereas a contract is a document containing promises by the contracting parties to each other (in some cases for the benefit also of third parties), a patent is a document which describes and claims an invention for the purposes of establishing a legal monopoly with regard to that invention. One cannot rationally interpret a patent claim without taking these matters into account. Moreover, I do not consider that Lord Neuberger can have meant anything different, even though he appears to have eschewed the expression "purposive construction" when describing the correct approach. On the contrary, in the passages relied upon by counsel for the Claimants, he expressly stated that a patent was to be interpreted through the eyes of the person skilled in the art and that the exercise involved interpreting the words of the claim in context. The context must include the very purpose for which the document exists, namely to describe and claim an invention.
- I would like to emphasise, for the avoidance of doubt, that I have not transposed the identities of the parties in the preceding discussion. The relevance of this statement will become apparent in the next section of this judgment.
Claim 1
- There is a dispute as to what is meant by the words "for use in treating a patient who is suffering from a relapsing form of [MS]" in claim 1. The Claimants contend that this feature of the claim merely requires that the 40 mg TIW regimen be suitable for treating such a patient, that is to say, that it has some degree of efficacy. The Defendants contend that it requires that the 40 mg TIW regimen reduces the annualised relapse rate by about 30%, that is to say, to be essentially equivalent in efficacy to the 20 mg QD regimen.
- In my judgment the Claimants' construction is the correct one. The Defendants' construction involves reading in words which are not present in the claim. The Defendants rely upon the facts that the primary endpoint defined in the specification at [0109] is a 30% reduction in relapse rate compared to placebo and that the skilled person would be well aware that the 20 mg QD regimen achieved a 30% reduction. As the Claimants point out, however, that is merely the hoped-for outcome of the proposed Phase III trial. Moreover, the specification expressly teaches the skilled reader at [0078] that the example is non-limiting. There is nothing in the specification to suggest that the patentee intended to limit the claim to a regimen which achieved a 30% reduction. As the skilled reader would appreciate, a regimen which achieved a reduction of, say, 20% compared to placebo might well be of value, particularly if it had advantages in terms of tolerability.
- Furthermore, another difficulty with the Defendants' interpretation is that the claim extends to treatment of CIS. What is the comparator then? A reduction in annualised relapse rate does not make sense in that case. If the risk of progression is the comparator, the PreCISe trial demonstrated that 20 mg GA QD reduced the risk of developing clinically definite MS by 45%.
- Finally, the claim is not limited to a concentration of 40 mg/ml, but extends to other concentrations, in particular 40 mg/0.75 ml. It was Prof Zajicek's evidence that the skilled person would not know what the efficacy of a different concentration might be. Moreover, counsel for the Defendants put an FDA document (dated 3 January 2011, but not suggested to represent a different approach to that which would have been taken in August 2009) to Dr Green in cross-examination indicating that it was necessary to be cautious about "the possible impact of a higher concentration/lower volume formulation on the safety and efficacy of the product".
Claim 3
- It is common ground that the skilled person would interpret the words "wherein the tolerability of [GA]
is increased by reducing the frequency of an [IPIR] or an [ISR]" to require that the 40 mg TIW regimen reduce the frequency of IPIRs or ISRs compared to the 20 mg QD regimen. Unlike claim 1, the wording of claim 3 expressly requires a comparison to be made. Reading the specification, the skilled person would conclude that the intended comparison must be with the 20 mg QD regimen.
The prior art
- The Claimants rely upon three items of prior art.
Pinchasi
- Pinchasi is an International Patent Application filed by Teva entitled "Method of treating multiple sclerosis" published on 19 July 2007. Pinchasi states at page 4 lines 10-14 that "disclosed herein is the finding that administration of glatiramer acetate at a dose of 40 mg/day significantly improves efficacy but does not have a corresponding increase in adverse reactions suffered by the patient". Pinchasi goes on to disclose and claim a method of alleviating a symptom of a patient suffering from a relapsing form of MS which comprises periodically administering 40 mg GA by subcutaneous injection. In one embodiment, the periodic administration is daily. In another embodiment disclosed at page 8 lines 10-11 and claimed in claim 3, "the periodic administration is every other day." Nothing further is disclosed regarding the QOD embodiment as distinct from the QD embodiment.
- Example 1 of Pinchasi describes and presents the results of a randomised, double-blind clinical trial which investigated the efficacy and tolerability of a 40 mg QD regimen compared to the 20 mg QD regimen in 90 patients with RRMS. It is common ground that the skilled reader would appreciate that the clinical trial described in Pinchasi appears to be the same clinical trial as that reported in Cohen 2007 (i.e. the FORTE Phase II trial) even though there is no coincidence in authorship between the two documents. The skilled reader would note, however, that there are differences in the level of experimental detail provided, the presentation of the data and, in particular, the discussion between Cohen 2007 and Pinchasi, with more information in Cohen 2007. (To give a simple example, Cohen 2007 contains 19 references whereas Pinchasi contains none.) I do not understand it to be in dispute, but in any event I find, that, to the extent that they differ, the skilled person would regard the interpretation of the data in Cohen 2007 as more reliable because it was more detailed and had been peer-reviewed.
- Apart from the references to the 40 mg QOD embodiment, the Claimants particularly rely upon two passages in Pinchasi. The first is the results section, where the specification states at page 15 lines 21-22:
"The safety profile of the 40 mg/d dose is essentially similar to the currently available 20 mg/day dose with a slight tendency of higher incidence of IPIR."
- The skilled reader would note, however, that the data in Table 4 shows that most types of IPIR increased in frequency with the 40 mg dose (from 22.7% to 32.6% for any symptom), although the data in Table 5 shows that ISRs slightly decreased in frequency (from 86.4% to 84.8%).
- The second passage is in the conclusions section where the specification states at page 19 lines 8 to page 20 line 6:
"The increased efficacy observed with 40 mg/day GA in reducing MRI-measured disease activity and relapse rates indicates that it is well tolerated and can improve the treatment of RRMS patients. The improvement in efficacy, however, is not accompanied by a corresponding increase of adverse reactions which would be expected upon a doubling of the administered dose.
Also observed was the accelerated rate at which the 40 mg/day dose became effective as compared to the 20 mg/day dose. This was unexpected. Specifically, the 40 mg/day dose showed efficacy, as measured by MRI, by the third month, whereas the 20 mg/day dose did not show efficacy until the sixth month. The results at three months comparing the 40 mg/day dosage are shown in Figure 3 and Table 2 above."
- The skilled reader would note, however, that the trial failed its primary endpoint, in that it failed to show a statistically significant difference in efficacy between the 40 mg dose and 20 mg dose (although the total number of lesions at months 7, 8 and 9 was reduced by 38% in the 40 mg group compared to the 20 mg group, the p value was 0.0898, well above the conventional threshold for statistical significance of 0.05). The skilled reader would also note that, although table 2 showed a statistically significant difference of 48% at three months, this was a post-hoc analysis.
Caon
- Caon is an abstract by Caon et al, "Randomised, Prospective, Rater-Blinded, Four Year Pilot Study to Compare the Effect of Daily Versus Every Other Day Glatiramer Acetate 20mg Subcutaneous Injections in RRMS" from the 2009 American Academy of Neurology conference that was published in Neurology, 72, Suppl 3 on 17 March 2009. It is sufficiently brief for me to set it out in full:
"OBJECTIVE: We conducted a pilot trial to compare the effect of GA 20 mg SC daily to every other day (QOD) on clinical, MRI, and immunologic outcomes in RRMS. BACKGROUND: The recommended dose of glatiramer acetate (GA) for the treatment of RRMS is 20 mg SC daily though the optimal dose remains unknown. Recent studies failed to show improved efficacy with higher than the recommended dose. DESIGN/METHODS: Treatment naïve RRMS patients were randomized to GA 20 mg SC QD or QOD and followed prospectively for 2 years. After 2 years, patients in each group were given the option to continue or switch to the other group, and followed for an additional 2 years. EDSS was recorded every 6 months by a masked-rater. Brain MRI scans were obtained at baseline, years 2 and 4. Blood for immunologic testing was obtained at baseline and multiple time points after randomization. RESULTS: 30 patients were randomized to GA 20 mg SC given QD or QOD. Both groups were matched for age, disease duration, EDSS, relapse rate, and T2W lesions. After 2 years, there were no difference in the relapse rate, disease progression, or any MRI outcome. In vitro GA-proliferative responses and Th1/Th2 cytokine expression did not differ between the two groups at any time point after randomization. After 2 years, all patients in the QD group opted to switch to QOD. After a total of 4 years of prospective follow-up, there was no difference in the clinical efficacy between the 'QD-QOD cross over' and the 'always QOD' groups. Injection related lipoatrophy was significantly less in the QOD group. CONCLUSIONS/RELEVANCE: This pilot study suggests that GA 20 mg SC administered QD or QOD may be equally effective in RRMS. Larger, multi-center studies are warranted to confirm our findings and identifying the optimal dose of GA in RRMS."
- It can be seen that Caon is very similar in content to Khan 2008 (paragraph 131 above) and appears to relate to the same study.
Flechter
- Flechter is an International Patent Application filed by Yeda entitled "Alternate day administration of Copolymer 1 for treating autoimmune diseases" which was published on 13 April 2000. Flechter discloses the results of an open-label study in which 68 patients with RRMS or RPMS were given 20 mg Copolymer 1 (i.e. GA) every other day and monitored over two years. It is common ground that the skilled reader would appreciate that this appears to be the same trial as that described in Flechter 2002B.
- Flechter states at page 17 lines 13-27 that the results of the study indicate that:
"1. Treatment with 20mg Cop 1 in alternate day injections subcutaneously is safe and well tolerated.
2. Relapse rate decreases significantly and stabilises at lower levels.
3. Patients' disability, as measured by EDSS, does not deteriorate during the first year of treatment, although deterioration is observed during the second year. It may very well be that the natural history of the disease is attenuated by Cop 1.
4. The results obtained in this open-label study, with alternate day injection of Cop 1, showed safety and efficacy as well, as the daily injection studies reported in several controlled trials."
- Amongst Flechter's claims are claim 7, which is the use of GA for MS, and claim 8, which specifies the therapeutically effective amount as 1.0 mg to 40.0 mg.
Novelty
The law
- The House of Lords held in Synthon BV v SmithKline Beecham plc [2005], [2006] RPC 10 that it was necessary to satisfy two requirements in order to establish that a claim lacked novelty over a prior publication (whether written, oral or by use). First, the prior publication must disclose the claimed invention (prior disclosure). Secondly, the prior publication must enable the skilled person to perform the claimed invention using his common general knowledge (enablement). No issue arises with respect to enablement in this case.
- Lord Hoffmann, with whom the other members of the House of Lords agreed, summarised the requirement for prior disclosure as follows:
"22.
the matter relied upon as prior art must disclose subject-matter which, if performed, would necessarily result in an infringement of the patent. That may be because the prior art discloses the same invention. In that case there will be no question that performance of the earlier invention would infringe and usually it will be apparent to someone who is aware of both the prior art and the patent that it will do so. But patent infringement does not require that one should be aware that one is infringing
It follows that, whether or not it would be apparent to anyone at the time, whenever subject-matter described in the prior disclosure is capable of being performed and is such that, if performed, it must result in the patent being infringed, the disclosure condition is satisfied.
24. Although it is sometimes said that there are two forms of anticipatory disclosure: a disclosure of the patented invention itself and a disclosure of an invention which, if performed, would necessarily infringe the patented invention
they are both aspects of a single principle, namely that anticipation requires prior disclosure of subject-matter which, when performed, must necessarily infringe the patented invention."
- Prior to 12 July 2017 it was settled law that the claim should be interpreted in the same manner, and had the same scope, for the purposes of considering both novelty and infringement: see Terrell on the Law of Patents (18th ed, 2016) at §§9-12 to 9-19 and the authorities cited, many of which emphasise the importance of ensuring that the patentee cannot maintain a broad scope of claim for the purposes of infringement, but a narrow one for the purposes of validity.
- In Actavis v Lilly the Supreme Court held that a claim could be infringed on the basis of a doctrine of equivalents even though the allegedly infringing act did not fall within the claim on its proper interpretation. This requires the court to answer the three questions set out in the judgment of Lord Neuberger at [66]:
"i) Notwithstanding that it is not within the literal meaning of the relevant claim(s) of the patent, does the variant achieve substantially the same result in substantially the same way as the invention, ie the inventive concept revealed by the patent?
ii) Would it be obvious to the person skilled in the art, reading the patent at the priority date, but knowing that the variant achieves substantially the same result as the invention, that it does so in substantially the same way as the invention?
iii) Would such a reader of the patent have concluded that the patentee nonetheless intended that strict compliance with the literal meaning of the relevant claim(s) of the patent was an essential requirement of the invention?"
If questions (i) and (ii) are answered yes, and question (iii) is answered no, then there is infringement.
- Another question which arises in the light of the Supreme Court's decision is what effect, if any, this has on the law of novelty. This issue is not addressed in the judgment of Lord Neuberger, and he makes no reference to Synthon v SKB. It will require another decision of the Supreme Court to supply a definitive answer to the question. Given the conclusion I have come to with respect to obviousness, however, I do not propose to consider the matter at length.
- Counsel for the Claimants submitted that it remained the law that a claim lacked novelty if the prior publication disclosed subject-matter which, if performed, would necessarily infringe the claim. Even if the subject-matter would not fall within the claim on its proper interpretation, it was sufficient that the subject-matter would infringe the claim applying the doctrine of equivalents. Otherwise, a claim could be infringed by a person who did exactly what the prior publication taught, yet the claim would be novel over that prior publication. This would be a radical departure from English patent law as it had previously been understood for many decades.
- Counsel for the Defendants submitted that it was no longer the law that a claim lacked novelty if the prior publication disclosed subject-matter which, if performed, would necessarily infringe the claim. Rather, the claim would only lack novelty if the prior publication disclosed subject-matter which fell within the claim on its proper interpretation. It was not sufficient that the subject-matter would infringe the claim applying the doctrine of equivalents.
- In support of this submission, counsel for the Defendants made three main points. The first is that in Synthon v SKB the House of Lords had not been considering the question of anticipation by equivalents, because at that time it was not possible to infringe by virtue of a doctrine of equivalents if the alleged infringement fell outside the claim on its proper interpretation.
- The second point is that it is established in the jurisprudence of the Boards of Appeal of the EPO that a claim is not deprived of novelty by an obvious equivalent of a feature in a prior publication. Thus the Case Law of the Boards of Appeal of the European Patent Office (8th ed, 2016) states at pages 108-109:
"4.5. Taking equivalents into account
The case law of the boards of appeal is based on a narrow concept of novelty, i.e. the disclosure of a prior document does not include equivalents of the features which are explicitly or implicitly disclosed; equivalents can only be taken into account when it comes to considering inventive step (T 517/90). This narrow concept of novelty, which excludes equivalents, is of particular importance for the application of Art. 54(3) EPC. In T 167/84 (OJ 1987, 369) the board commented that conflicting applications within the meaning of Art. 54(3) EPC 1973 were included in the state of the art solely from the point of view of novelty, but were considered in the light of their 'whole contents'. In order to mitigate the harsh effects of the 'whole contents approach', its application was confined to novelty. Further, in order to reduce the risk of 'self-collision', it had always been considered justified to adopt a strict approach to novelty. For this reason, the Guidelines expressly stated that 'when considering novelty, it is not correct to interpret the teaching of a document as embracing well-known equivalents which are not disclosed in the document; this is a matter of obviousness' (Guidelines G-VI, 2 November 2015 version). According to the case law of the boards of appeal the 'whole contents' of an earlier document did not also comprise features which were equivalents of features in the later document (see also T 928/93, T 1387/06).
In T 652/01 the appellant was of the opinion that although the relevant prior-art document did not explicitly mention a particular feature, that feature could be derived from the document by applying the document's teaching mutatis mutandis. The appellant had referred to T952/92 (OJ1995,755), which, in its first headnote, stated that 'availability' within the meaning of Art. 54(2) EPC 1973 involved not only availability of the disclosure but also availability of information accessible and derivable from the disclosure, which meant that 'derivable equivalents' were included. However, the board held that, when reading the cited phrase from T 952/92 in the context of the present decision, it was clear that the term 'derivable' had been employed in the sense of "obtainable by chemical analysis of a sample" and that it was used with the same restriction as expressed in opinion G 1/92 (OJ 1993, 277), namely that it had to be 'directly and unambiguously derivable'."
- The third point is that the decision of the Supreme Court was based on Article 2 of the Protocol on the Interpretation of Article 69 of the European Patent Convention, which is concerned with the extent of protection conferred by a European patent or patent application, that is to say, with infringement and not with validity.
- The conclusion I reach is that counsel for the Defendants is correct. Nevertheless, in case this case goes further, I shall consider what the position would be if counsel for the Claimants were correct.
Novelty over Pinchasi
- Claim 1. It is common ground that:
i) Pinchasi teaches administration of 40 mg GA QOD to treat patients with relapsing forms of MS;
ii) over a seven-day period, that regimen would be identical to the regimen claimed in the Patent; and
iii) over a longer period, that regimen would be identical to the regimen claimed in the Patent every other week, while in the intervening weeks it would result in the administration of one additional dose.
- The Claimants contend that this is sufficient to deprive claim 1 of novelty even before one comes to any question of equivalents. I do not accept this. MS is a chronic disease which requires long-term treatment with DMTs such as GA. It is clear from the evidence that, at least in this context, the skilled person would regard QOD and TIW as distinct regimens. Furthermore, they result in the administration of different quantities of GA over any period longer than one week. Still further, as the Claimants themselves contend, TIW has the advantage that it enables patients to have fixed-day administration and injection-free weekends if they wish, whereas QOD does not.
- In the alternative, the Claimants contend that Pinchasi deprives claim 1 of novelty because claim 1 would be infringed by the administration of 40 mg QOD applying the doctrine of equivalents. For this purpose, it must be assumed that the skilled person would consider it plausible that 40 mg TIW is efficacious as claimed by the Patent (as to which, see below). Counsel for the Claimants relied upon the evidence of Dr Green and Prof Zajicek as establishing that the answers to questions (i) and (ii) were both yes, because the skilled person would think that missing one dose every fortnight was unlikely to have a detrimental effect on the efficacy of the treatment. Counsel for the Defendants submitted that it had not been established that 40 mg QOD was equivalent to 40 mg TIW because this would require a clinical trial, which had not been carried out. In the absence of a clinical trial, however, I have to determine the issue on the balance of probabilities on the evidence that is before the court. Accordingly, I conclude that questions (i) and (ii) should both be answered yes.
- Turning to question (iii), counsel for the Claimants submitted that the skilled reader would not have concluded that strict compliance with the literal meaning of this feature of claim 1 was required. Counsel for the Defendants submitted that the skilled reader would have reached the opposite conclusion for two reasons. First, because that was necessary to avoid anticipation by Pinchasi. I do not accept this, since there is no mention of Pinchasi in the Patent. Secondly, because TIW has the advantage of enabling patients to have fixed-day administration and injection-free weekends as discussed above. I do not accept this either, since there is no suggestion that all patients would regard that as advantageous nor is there any evidence that it would make a major difference to those patients who did regard it as advantageous. More generally, I do not consider that there is anything in the specification to suggest to the skilled reader that strict compliance with the literal meaning of this feature of claim 1 was required.
- Accordingly, I conclude that, if it is legally possible for a claim to be deprived of novelty by virtue of the doctrine of equivalents, then claim 1 lacks novelty over Pinchasi.
- Claim 3. Counsel for the Claimants submitted that, if claim 1 lacked novelty, then so did claim 3 since administering 40 mg QOD would inevitably result in lower frequency of ISRs and/or IPIRs compared to 20 mg QD. Counsel for the Defendants did not dispute this.
Obviousness over the prior art
The law
- The structured approach to the assessment of allegations of obviousness first articulated by the Court of Appeal in Windsurfing International Inc v Tabur Marine (Great Britain) Ltd [1985] RPC 59 was re-stated by Jacob LJ in Pozzoli v BDMO SA [2007] EWCA Civ 588, [2007] FSR 37 at [23]. The correct approach to the fourth step was summarised by Kitchin LJ in MedImmune Ltd v Novartis Pharmaceuticals Ltd [2012] EWCA Civ 1234, [2013] RPC 27 at [90]-[93].
Obviousness over Pinchasi
- Claim 1. The sole difference between Pinchasi and claim 1 of the Patent is that Pinchasi discloses a regimen of 40 mg QOD while claim 1 requires a regimen of 40 mg TIW. As discussed above, the difference amounts to one dose every fortnight.
- Counsel for the Claimants put the Claimants' case that claim 1 was obvious in two ways which I will consider in turn. Again, it must be assumed for these purposes that the skilled person would consider it plausible that 40 mg TIW is efficacious as claimed by the Patent
- First, he argued that 40 mg TIW was an obvious alternative to 40 mg QOD for the same reasons as he relied upon in support of his argument on anticipation by equivalence: the skilled person would think that missing one dose a fortnight was unlikely to have a detrimental effect on the efficacy of the treatment, and TIW would have the advantages that it was likely to increase the tolerability of the medication and would be more convenient for patients who wanted fixed-day administration and weekends injection-free. Moreover, the skilled person would know that Rebif was administered TIW. In other words, 40 mg TIW was nothing more than a small and simple variation on Pinchasi's teaching. I accept this argument.
- Secondly, counsel for the Claimants argued that, if the skilled person considered the matter in more detail in the light of his common general knowledge and his literature search, then 40 mg TIW would be an obvious regimen to try in a Phase III clinical trial and the skilled person would have a fair expectation of success, both in the sense that the regimen would be efficacious compared to placebo and in the sense that it would be comparable in efficacy to 20 mg QD. Counsel for the Claimants submitted that this was supported by the evidence of both Dr Green and Prof Zajicek.
- As to this, the Defendants contend that it was not obvious to try 40 mg TIW, for a combination of three reasons. First, the Defendants contend that the skilled person would not have been interested in improving the administration of GA, because of the new treatments on the horizon. I do not accept this. The new treatments were still in the future. In August 2009 many, many patients worldwide were receiving GA. After the CORAL trial, the only real option so far as GA was concerned was to try to reduce the frequency of injections. Prof Zajicek accepted that there was an unmet need for a less frequent administration in August 2009, at least if it matched the efficacy and reduced the side effects compared to 20 mg QD.
- Secondly, the Defendants contend that the skilled person would not be motivated to consider a 40 mg TIW regimen. If the skilled person was interested in reducing the frequency of administration, then the obvious regimen to consider would be 20 mg QOD as suggested by the Fletcher 2002 and Khan 2008 publications. Again, I do not accept this. The skilled person had a clear motivation to reduce the frequency of administration of GA in order to increase tolerability, adherence and convenience. Moreover, a TIW regimen had the advantages of fixed-day administration and injection-free weekends, as already discussed. I agree that 20 mg QOD would be one obvious regimen to consider, but it does not follow that there were not other obvious regimens to consider. Pinchasi had proposed 40 mg QOD, and 40 mg TIW was another obvious regimen to consider.
- Thirdly, and most importantly, the Defendants contend that the skilled person would be dissuaded from trying a 40 mg dose because of the increase in the severity of ISRs and IPIRs reported in Cohen 2007 and Comi 2008B, and in particular the increase in early terminations due to adverse events mainly due to ISRs which had been reported in Comi 2008B, given that it had been found that 40 mg QD was no more efficacious than 20 mg QD. The principal evidence which the Defendants rely upon in support of this contention is the first sentence of paragraph 8.6 of Prof Zajicek's first report, which in turn relies upon Cohen 2007 (and not, it may be noted, Comi 2008B). When this was put to Dr Green, his opinion was that the skilled person might be concerned that increasing the dose from 20 mg to 40 mg would lead to an increase in ISRs, but would not be overly concerned. He disagreed that the skilled person would be dissuaded from trying 40 mg, in particular because the skilled person would expect that a reduction in the frequency of administration from QD from TIW would reduce the incidence of ISRs and IPIRs. Counsel for the Defendants submitted that counsel for the Claimants had not challenged Prof Zajicek's evidence on this point in cross-examination. I disagree with this. While it may be accurate that the specific passage was not explicitly challenged, Prof Zajicek's assessment of Cohen 2007 (and Comi 2008B for that matter) was challenged, as was his reasoning. In particular, it was put to Prof Zajicek, and he accepted, that the skilled person would expect that reducing the frequency of administration of 40 mg from QD to TIW would reduce the incidence of ISRs. It was not expressly put that the skilled person would also expect this to reduce the severity of ISRs, but there was no disagreement between the experts that the severity of ISRs was linked to their frequency. In any event, the law is clear is that I should not surrender my judgment to any witness. Given that Prof Comi's overall conclusion in Comi 2008B was that 40 mg QD was safe and well tolerated, and given that a reduction in the frequency of administration from QD to TIW would be expected to reduce the incidence of ISRs (and IPIRs), I consider that the skilled person would not be dissuaded from trying 40 mg TIW in a Phase III trial.
- The Defendants also contend that the skilled person would not have a fair expectation of success because he would be unable to predict the overall effect of increasing the dose from 20 mg to 40 mg and reducing the frequency of administration from QD to TIW. As to this, Dr Green agreed that the skilled person could not specifically predict the outcome, which is why the skilled person would conduct a trial. Dr Green's view, however, was that the skilled person would consider it likely that the efficacy of 40 mg TIW would be comparable that of 20 mg QD, bearing in mind that the total weekly dose would be similar (120 mg compared to 140 mg), and highly likely that 40 mg TIW would be efficacious compared to placebo. For his part, Prof Zajicek accepted that the skilled person would know that 40 mg TIW was potentially as efficacious as 20 mg QD. Accordingly, I conclude that the skilled person would have a strong expectation that 40 mg TIW would be efficacious compared to placebo and a reasonable expectation that it would be comparable in efficacy to 20 mg QD.
- For completeness, I will mention two additional factors which were relied on by Dr Green in support of his opinion that 40 mg TIW was obvious in the light of Pinchasi. The first was that the skilled person would expect the effects of GA to last longer than 24 hours. I have not found that to have been common general knowledge, however. The second was the possibility suggested by the FORTE trials that the 40 mg dose might have an earlier onset of action than 20 mg dose and that this might be clinically beneficial. I agree that, if the skilled person took this factor into account, it would encourage him to try 40 mg TIW and to expect success rather than discourage him, but in my assessment the skilled person would place little weight on this factor.
- Overall, I accept the Claimants' case that 40 mg TIW was obvious to try and that the skilled person would have had a fair expectation of success, both in the sense that the regimen would be efficacious compared to placebo and in the sense that it would be comparable in efficacy to 20 mg QD. It follows from this conclusion that claim 1 would be obvious even if it were construed in the manner contended for by the Defendants.
- Claim 3. Counsel for the Claimants submitted that, if claim 1 was obvious over Pinchasi, then so was claim 3. Counsel for the Defendants did not dispute this.
- Secondary evidence. The Claimants rely upon two matters by way of secondary evidence to support the contention that the claimed inventions were obvious.
- First, the Claimants rely upon the contents of a number of internal Teva documents which were identified in the first instance through discovery in the US proceedings and were then ordered to be disclosed in these proceedings. The Claimants contend that what these documents show is that the applications for the Patents and its counterparts were filed by Teva as an exercise in "life-cycle management" of Copaxone when faced within the prospect of the future expiry of 888 and consequent generic competition in the 20 mg QD market, that the 40 mg TIW regimen was arbitrarily chosen from a number of possible alternatives and that the decision-making was driven by Teva's management rather than by the named inventor Ety Klinger. The Defendants dispute that this is what the documents show. In any event, the question of whether the claimed inventions are obvious over the prior art is an objective question to be assessed from the perspective of the person skilled in the art in light of his common general knowledge. An invention devised for commercial reasons may nevertheless not have been obvious judged by that standard, just as an invention which the named inventor believed to be ground-breaking may in fact have been obvious.
- Secondly, the Claimants rely upon a proposal filed by Teva with the FDA on 17 December 2009 for a multi-centre, randomised, double-blind Phase III clinical trial to compare the efficacy, safety and tolerability of GA 40 mg TIW compared to placebo in patients with RRMS (the Glatiramer Acetate Low frequency Administration or GALA trial, the results of which were subsequently reported in Khan et al, "Three times weekly glatiramer acetate in relapsing-remitting multiple sclerosis", Ann. Neurol., 73(6), 7050123 (2013))). Section 3.3.2 of the proposal headed "Rationale for Dose Regimen Selection" states (emphases added):
"The accumulated data showing that GA acts as an immunomodulating drug, and the results of the FORTE study showing that a higher dose of GA does not increase the efficacy of the drug, encouraged the Sponsor to further explore the optimal dose and dosing regimen of GA and to study the safety and efficacy of GA administered in a reduced injection frequency. This rationale is further supported by the current situation where all the currently approved treatments for RRMS are administered parenterally and frequent injections affect the long-term adherence to treatment.20
Hence, the next natural step is to reduce the dosing regimen of GA and find the optimal regimen that will improve the convenience of treatment and reduce the burden and adverse events associated with daily subcutaneous injections.
Further support for this rationale was provided by several small-scale, investigator initiated studies, which explored a reduced injection frequency of GA.21,22,23 These studies employed a dose of GA 20mg, given either on alternate days or twice weekly, with follow-up periods which ranged from two to four years. These studies included between 30 and 68 subjects, and employed an open label design. The results of these studies demonstrated effects in relapse rate reduction which were comparable to daily injections of GA 20mg, suggesting a lower injection frequency can be considered. In two of these studies,22, 23 these comparable effects were also supported by MRI data. These results of these studies also suggest that participating subjects found the reduced frequency regimens preferable and associated with a lower of injection related adverse events. The authors of all three studies highlighted the need for larger, multi-center studies to confirm their findings.
While it is important to note that all the above mentioned studies were clearly too small and underpowered to show a significant effect on clinical end-points, they do provide additional support to the rationale of the current proposed study.
The dose regimen that was selected for the trial is GA 40 mg injected subcutaneously three times a week.
In the absence of data about the efficacy of lower frequency administration of GA 20mg in large, well controlled studies, the sponsor has decided to take a more conservative approach. Therefore the subjects will receive approximately the same weekly dose, given by 3 subcutaneous injections instead of with a daily injection frequency of 7 injections.
The 40 mg dose has been previously explored as a potential formulation for daily injection in two clinical studies (see Section 3.2.2.2). While no superiority of the GA 40 mg dose over the GA 20 mg was demonstrated, the GA 40 mg formulation showed similar quality, safety and efficacy profile compared to that of the GA 20 mg dose. The use of the 40mg dose in this study would allow a gradual approach where reduction in injection frequency would be offset by the use of a higher dose, already shown to be effective and safe in daily use.
Three weekly doses of 40 mg GA would give a cumulative weekly dose of 120mg, approximately similar to the 140 mg cumulative weekly dose given today with daily injections of 20 mg. This would serve as an opportunity to explore the increased convenience of three injections a week, without substantially reducing the total GA dose administered. If this dose regimen is indeed found to be effective, RRMS patients will benefit from a much more convenient injection schedule, and the enhanced compliance resulting from this will help maintain the long term efficacy of this treatment.
It should also be noted that considering the reduced injection frequency, and the fact that in the GA/9016 study, local reactions at the injection sites and immediate transient reactions following injection were reported with a similar incidence for both doses,15 one may certainly expect a reduction in the frequency of such reactions with this new dose regimen, further enhancing subject adherence to treatment."
References 21 and 22 are Flechter 2002B and Khan 2008. (Reference 23 is another abstract from Prof Khan's group comparing 20 mg QD with 20 mg twice-weekly published in 2009, I presume after the priority date of the Patent. It is not suggested, however, that this would have affected the skilled person's attitude to the proposal.)
- The Claimants contend that this proposal, and its acceptance by all the clinical investigators who enrolled patients in the GALA trial in 142 sites in 17 countries, supports their case that 40 mg TIW was obvious to try and that the skilled person would have had a fair expectation of success, and accordingly that claims 1 and 3 of the Patent were obvious. I agree with this.
Caon and Flechter
- In view of the conclusions reached above, I can take these shortly. In essence, both Caon and Fletcher disclose the administration of 20 mg GA QOD. In both cases the results suggest that 20 mg QOD may be as efficacious as 20 mg QD, but the evidence is weak due to the nature of the trials. Given my conclusion that claims 1 and 3 of the Patent are both obvious over Pinchasi, it is unnecessary to decide whether they are obvious over Caon and Flechter. If the claims are not obvious over Pinchasi, then in my view they cannot be obvious over either Caon or Flechter.
Lack of inventive step for want of technical contribution and insufficiency
- The Claimants contend that, if the claimed inventions were not obvious over the prior art, then there is nothing in the Patent which makes it plausible that 40 mg TIW would be efficacious compared to placebo or as efficacious as 20 mg QD or more tolerable, because the Patent contains neither any clinical data nor any theoretical justification for such a supposition beyond what was already known to the skilled person. Accordingly, the Claimants contend that the claimed inventions make no technical contribution to the art and are therefore do not involve an inventive step in accordance with the principles summarised by Floyd LJ in Generics (UK) Ltd v Yeda Research and Development Co Ltd [2013] EWCA Civ 925, [2014] RPC at [49], alternatively are insufficient in accordance with the principles summarised by Kitchin LJ in Idenix Pharmaceuticals Inc v Gilead Sciences Inc [2016] EWCA Civ 1089 at [137]-[138]. The principles applied by the English courts are based on those developed by the Boards of Appeal of the EPO, which are well illustrated by two recent contrasting decisions of the same Board relating to dasatinib in Cases T 488/16 (1 February 2017) and T 950/13 (3 February 2017).
- This contention is advanced by the Claimants by way of a squeeze. Counsel for the Defendants submitted that there was no squeeze, however, because the legal tests to be applied were different: obviousness required a fair expectation of success, whereas plausibility was a lower standard which merely required that the claim should not be speculative. In support of this submission, he cited Generics (UK) Ltd v Warner-Lambert Co LLC [2016] EWCA Civ 1006, [2017] RPC 1 at [133] (Floyd LJ) and Actavis Group PTC EHF v Eli Lilly and Co [2015] EWHC 3294 (Pat), [2016] RPC 12 at [177] (Henry Carr J).
- Counsel for the Claimants accepted that the legal tests to be applied were different. He submitted that this did not matter on the facts of the present case because all that the Defendants could rely upon to establish that the claimed inventions were not speculative was the skilled person's common general knowledge outlined above. If 40 mg TIW was not obvious to try with a fair expectation of success in the light of the skilled person's common general knowledge, then the claim that 40 mg TIW would be efficacious compared to placebo was speculative and the claim that it would be as efficacious and more tolerable than 20 mg QD was even more speculative.
- Counsel for the Defendants submitted that this argument failed to take into account the need to provide pharmaceutical companies with an incentive to finance the huge cost of Phase III trials and the need for pharmaceutical companies to file patent applications before the results of such trials were known if the applications were to have any real prospect of leading to valid patents. I acknowledge the relevance of these policy considerations, but in my view they are already reflected in the legal tests applied by the courts. As is well known, the patent system does not always provide a satisfactory incentive for innovation. The remedy for such problems lies elsewhere.
- In my judgment, if the claimed inventions were not obvious, then the claim that 40 mg TIW would be efficacious compared to placebo was credible, but the claim that it would be as efficacious and more tolerable than 20 mg QD was speculative. I therefore conclude that claim 3 would be invalid for lack of an inventive step and insufficiency, but not claim 1 as I have construed it.
- Counsel for the Claimants also contended in his closing submissions that the claims lacked an inventive step, alternatively were insufficient, because they covered concentrations other than 40 mg/ml, but the skilled person could not predict the efficacy or tolerability of other concentrations and thus, even if the claims were plausible with respect to 40 mg/ml, they were not plausible with respect to other concentrations. Counsel for the Defendants submitted that this contention was not open to the Claimants since it had not been pleaded. I agree with this.
- In case this case goes further, I must briefly address the Defendants' reliance upon evidence which post-dates the priority date of the Patent. It is common ground that such evidence can only be relied upon to confirm the existence of a technical effect which is plausible in the light of the specification and the skilled person's common general knowledge, and not to establish the existence of a technical effect for the first time.
- The Defendants rely upon Wolinsky et al, "GLACIER: An open-label, randomised, multicentre study to assess the safety and tolerability of glatiramer acetate 40mg three-times weekly versus 20mg daily in patients with relapsing-remitting multiple sclerosis", Multiple Sclerosis and Related Disorders, 4, 370-376 (2015). This reports the results of a 209 patient open-label study comparing the perceived treatment convenience and rates of injection related adverse events ("IRAEs", essentially ISRs plus IPIRs) for patients treated with GA 20 mg QD and 40 mg TIW. The authors concluded that the 40mg TIW group reported 50% fewer IRAEs, and a 60% reduction in the annualised rate of moderate/severe IRAEs, compared to the 20mg QD group.
- As Dr Green pointed out, the Patent does not teach the skilled person to undertake the GLACIER trial. In any event, it was common ground between Dr Green and Prof Zajicek that the methodology of the GLACIER trial was flawed in a number of respects. First, it was an open-label trial. Secondly, one of the secondary endpoints of the study was the patients' expectation of convenience, efficacy and safety of 40mg TIW compared to 20mg QD, which had the potential to introduce bias. Thirdly, although the rate of IRAEs was a primary endpoint of the trial, the severity of IRAEs was not a primary, secondary or additional endpoint, and the conclusion about the rate of moderate/severe events was based on post-hoc analysis.
- Accordingly, I conclude that the GLACIER trial adds little of any weight to what the skilled person would have expected in August 2009 based on their common general knowledge.
Arrow declaration
- In addition to revocation of the Patent, the Claimants seek a declaration that that the use of the Claimants' Product for treating a patient suffering from relapsing forms of MS wherein the Claimants' Product comprises glatiramer acetate for administration in a regimen of three subcutaneous injections of a 40mg dose of glatiramer acetate every seven days with at least one day between each subcutaneous injection would have lacked novelty and/or been obvious as at 20 August 2009 with regard to the dosing regimen. In support of this claim, the Claimants relied on a witness statement made by Jennifer Sunderland, Mylan's Senior Patent Litigation Counsel, which was unchallenged.
The law
- The relevant legal principles have been recently considered in the AbbVie litigation: see Fujifilm Kyowa Kirin Biologics Co Ltd v AbbVie Biotechnology Ltd [2017] EWCA Civ 1, [2017] RPC 9 (CA) and [2017] EWHC 395 (Pat) (Henry Carr J).
- The jurisdictional position was summarised by Floyd LJ delivering the judgment of the Court of Appeal at [98] as follows:
"
we do not consider that there is any issue of principle which prevents the granting of Arrow declarations in appropriate cases. Drawing the threads together:
(i) A declaration that a product, process or use was old or obvious at a particular date does not necessarily offend against s.74 of the Act.
(ii) Such a declaration may offend against the Act where it is a disguised attack on the validity of a granted patent.
(iii) Such declarations do not offend against the scheme of the EPC or the Act simply because the declaration is sought against the background of pending divisional applications by the counter-party.
(iv) On the other hand the existence of pending applications cannot itself be a sufficient justification for granting a declaration.
(v) Whether such a declaration is justified depends on whether a sufficient case can be made for the exercise of the court's discretion in accordance with established principles."
- The principles upon which the Court will grant discretionary relief were considered by Henry Carr J at [365]-[371]. In summary, the Court must consider:
i) justice to the claimant;
ii) justice to the defendant;
iii) whether the declaration will serve a useful purpose. The attainment of commercial certainty in patent cases can constitute a useful purpose. The spin-off value of a judgment in other states may be such a factor, but a declaration sought solely for the benefit of foreign courts will rarely be justified; and
iv) whether or not there are any other special reasons why the court should or should not grant the declaration.
Assessment
- The Patent stems from a divisional of European Patent 2 405 749 ("749"). On 2 February 2017 Teva withdrew the text of 749 before a Technical Board of Appeal could rule upon its validity in the light of (among other citations) Pinchasi. There is no dispute that Teva has filed two further pending divisional applications which cover the 40 mg TIW regimen for administration of GA, namely European Patent Applications Nos. 2 630 962 ("962A") and 3 199 172 ("172A"). It is for this reason that the Claimants seek an Arrow declaration in the following terms:
"A declaration that the use of the Claimants' Product for treating a patient suffering from relapsing forms of MS wherein the Claimants' Product comprises glatiramer acetate for administration in a regimen of three subcutaneous injections of a 40mg dose of glatiramer acetate every seven days with at least one day between each subcutaneous injection would have lacked novelty and/or been obvious as at 20 August 2009 insofar as the dosing regimen is concerned."
- In the light of my conclusions above, I consider that the use of the Claimants' Product in a 40 mg TIW regimen would have been novel, but obvious, as at 20 August 2009. That is a necessary, but not a sufficient, foundation for the relief sought by the Claimants. I turn, therefore, to consider the factors relied upon by the Claimants as justifying the grant of an Arrow declaration.
- First, the Claimants rely upon the pendency of 962A and 172A. It is clear from the judgment of the Court of Appeal in AbbVie, however, that the existence of pending divisional applications is not sufficient to justify the making of an Arrow declaration. More is required.
- Secondly, the Claimants contend that the Defendants have sought to shield the subject-matter of the Patent from scrutiny by the courts of this country and the Netherlands and the Boards of Appeal. I do not accept this. Whatever may have happened elsewhere in other cases, the Defendants have vigorously defended the validity of the Patent at trial before this Court, resulting in the giving of this judgment.
- Thirdly, the Claimants rely upon the spin-off value of a declaration even if the declaration is confined to the UK. When I asked counsel for the Claimants why an Arrow declaration should have any greater persuasive value than a reasoned judgment of this Court on the validity of the Patent, however, he had no real answer.
- Fourthly, the Claimants rely upon Teva's public statements to the effect that it intends to enforce its patent portfolio, including the Patent. In itself, however, there is nothing wrong with this. Moreover, in future, Teva will have to acknowledge the judgment of this Court (subject, of course, to any appeal).
- Fifthly, the Claimants rely upon their need for commercial certainty. I am unclear, however, how an Arrow declaration would provide the Claimants will greater certainty than this judgment. If this judgment stands, but 962A and/or 172A proceed to grant, then any claim for infringement by the Defendants against the Claimant in respect of the 40 mg TIW regimen can be met by an application for summary judgment seeking revocation of the patent(s) relying upon issue estoppels arising out of this judgment. An Arrow declaration would not preclude the need for an application for summary judgment, albeit based on the declaration, in such circumstances.
- Accordingly, I decline to grant the Claimants an Arrow declaration.
Summary of conclusions
- For the reasons given above, I conclude that:
i) claims 1 and 3 of the Patent are novel over Pinchasi;
ii) claims 1 and 3 of the Patent are obvious over Pinchasi, and therefore invalid;
iii) if claims 1 and 3 were not obvious over Pinchasi, then they would not be obvious over Caon or Flechter;
iv) if claim 3 was not obvious, then it would lack plausibility and thus be invalid on the grounds of lack of inventive step and insufficiency, but the same does not apply to claim 1 as I have construed it; and
v) it is not appropriate to grant the Claimants an Arrow declaration.
